单细胞eQTL分析 | 启航单细胞多组学大数据时代
发布时间:2022-11-03 10:03:37
——听说了吗?单细胞多组学最近有个大动作?
——是呀,它就是表达数量性状基因座(eQTL)分析,可以用于解析基因突变对基因表达水平的调控关系,一起来看看呀。
判断一个基因与某一特定疾病之间是否存在相关性的常用办法是利用存在于人类整个基因组的单核苷酸多态性(Single-nucleotide polymorphisms,SNPs)(图1),进行全基因组关联分析(Genome-wide association studies,GWASs)。如果一个SNP与一个基因位置靠近或者位于该基因内,而该基因上的某个SNP突变可能造成疾病,这个SNP与该致病突变将在人群中紧密连锁。基于此,可以在几千或者更多患者中提取DNA,并将这些DNA与大约相等数量的健康人群(最好是患者的健康亲属或同种族和地理分布相同的群体)中提取的DNA样品进行对比,寻找辨别出与疾病联系最密切的SNPs。
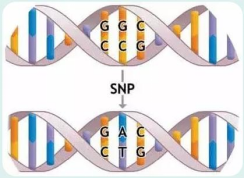
图1 单核苷酸多态性(SNP)
疾病相关的遗传学研究经常会发现一些致病基因或易感基因突变,但是这些突变与表型的关联有时并不十分直接,例如突变通过影响某个基因的表达量进行诱导疾病发生。而在测序过程中,发现的某个基因发生突变(如产生SNP变化),但这样的改变可能并不一定影响基因和蛋白的表达,也不会影响疾病或其他生物学过程。因此科学家想到可以利用mRNA序列来研究DNA的改变。表达数量性状基因座(Expression quantitative trait loci,eQTL)分析,是功能基因组学中广泛使用的工具,用于识别基因型与疾病联系和基因表达的遗传调控的机制(图2),其分析的核心在于获得单个DNA突变与单个基因表达量之间的相关性。

图2 eQTL是沟通基因改变与疾病的桥梁
eQTL分为顺式和反式两种:顺式eQTL(cis-eQTLs)是位于目标基因(eGene)(具有相关eQTL的基因)的距离截断(distance cutoff)内,例如位于目标基因上游或下游1Mb范围内,并与其表达相关的基因组序列变异体。除此之外,任何位于这个基因组窗口之外的eQTLs都被认为间接调节基因表达,并被认为是反式eQTLs(Trans-eQTL)。
——单细胞测序也能进行eQTL研究吗?
——可以啊,新格元专注于打造单细胞多组学平台,单细胞层面的eQTL研究,我们已经搞定咯!
单细胞测序在eQTL研究中的重要性
在以往的eQTL研究中,高通量RNA测序(RNA-seq)由于具有更好的特异性和敏感性,以及检测新转录本、剪接连接和等位基因特异性基因表达的能力,因此被广泛应用。但是近年来大规模平行单细胞RNA测序(scRNA-seq)已越来越多地用作bulk RNA-Seq的强大替代品。与bulk方法相比,scRNA-seq的主要优势是能够揭示复杂和稀有的细胞群,揭示基因与基因之间、细胞与细胞之间的调控关系,并跟踪发育中不同细胞谱系的轨迹。虽然在2009年scRNA-seq诞生时,第一个数据集仅包含8个细胞,但在如今典型实验中的细胞数量通常接近数万甚至数十万以上。
单细胞RNA测序表达谱分别包括细胞的异质性和每个细胞的表达变异性,而整体RNA测序是一个组织中所有细胞的平均值,细胞的异质性无法估计(图3)。此外scRNA-seq还允许估算单个细胞基因表达的可变性。
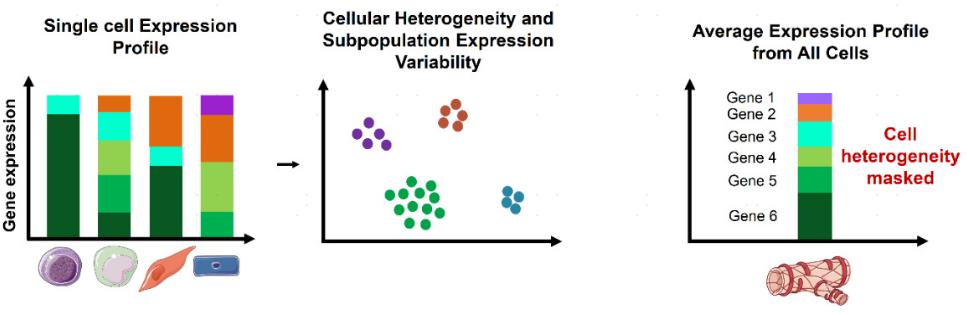
图3 相比bulkRNA测序,单细胞测定在分析不同细胞群体基因表达差异上具有明显优势[1]
从下图(图4)中顺式eQTL基因表达实例的小提琴图可知,与细胞类型显著相关的等位基因特异性表达信息在bulk RNA水平的分析中会被掩盖。因此相关的细胞类型特异性eQTL信息也会随之丢失。

图4 等位基因特异性基因在单个细胞类型中的表达(左图),但在大量组织分析中无法获得(右图)[1]
2013年,一项基于来自15个Hapmap淋巴母细胞系的1,440个细胞的研究已经证明了eQTLs的细胞类型特异性鉴定的概念,该研究使用高度平行的qPCR测量了92个基因的表达[2]。而首个真正意义上的单细胞层面的eQTL分析诞生于2018年,Kang等人利用液滴单细胞测序技术检测了从23个供体提取的8个主要免疫细胞群共78,000个PBMC细胞,并进行了eQTL分析[3]。该研究总共鉴定出32个cis-eQTL,其中22个具有细胞类型特异性。
同年,Van Derwijst等人的另一项研究使用scRNA-serq分析了来自生命线深度队列(Lifelines Deep cohort)的45名荷兰捐献者的25,000个PBMCs并进行了细胞类型特异性eQTLs鉴定(cell-type-specific eQTLs,ct-eQTL)[4]。作者将来自每个供体的相似细胞聚类成六个更广泛的分类:CD4+T细胞、CD8+T细胞、NK细胞、单核细胞、B细胞和DC[4],并与之前的两项大规模研究,即RNA-seq(n=2116)[5]和DeepSAGE(n=94)[6]进行了比较,发现分别有1%-8%的重叠(图5)。低重叠可以反映样本细胞组成的差异,即PBMCs与全血的差异,以及整体RNA-seq研究测序深度不够。他们发现了379个cis-eQTL(287个基因),其中331个(249个基因)在PBMC eQTLs(每个基因所有细胞的平均表达)体样分析中显著,48个(38个基因)只在特定细胞类型中检测到,而这些细胞类型特异性信号在bulk RNA-seq数据集中被稀释了。
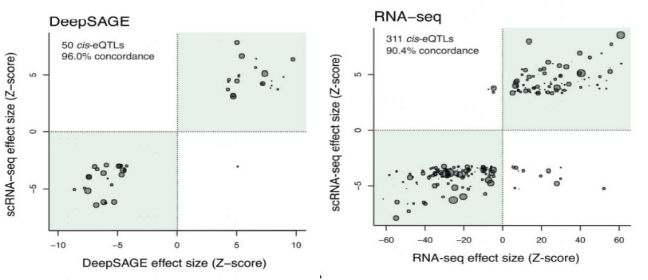
图5 DeepSAGE(左图)和RNA-seq(右图)获得的cis-eQTL数据量低于scRNA-seq
Van Derwijst等人的研究还强调了scRNA-seq的另一个优势是在单个供体的基础上确定共表达网络。他们证明,真正的基因相关性在他们的45个样本中比在数千个个体的大规模bulk RNA-Seq数据集中更明显(图6)。因此,细胞类型特异性的scRNA-seq数据也可用于识别改变共表达关系的SNPs,即共表达QTLs,并分析基因网络的方向性和上下游特异性[4]。
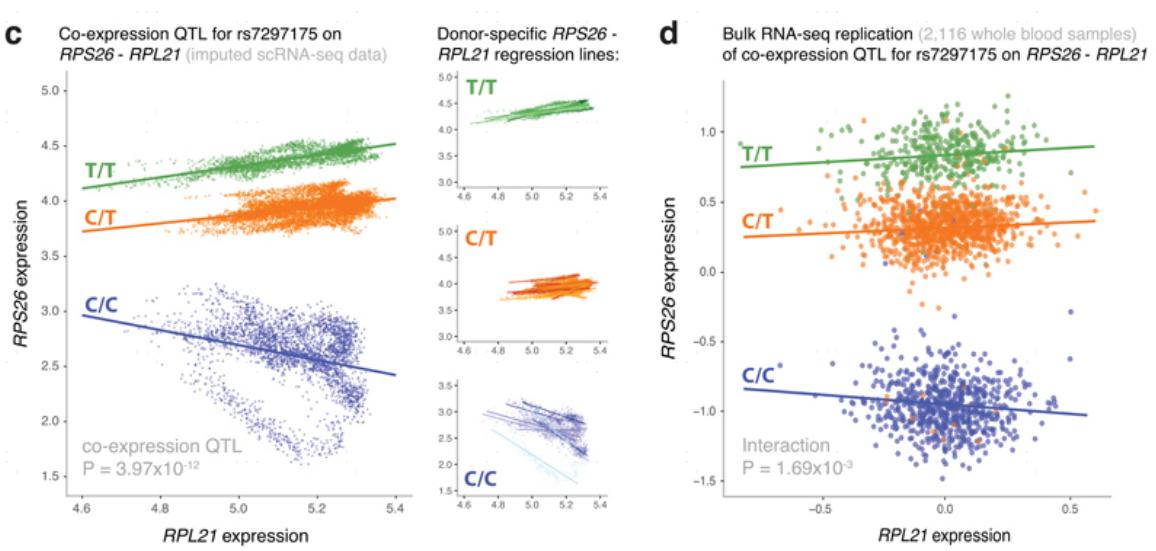
图6 scRNA-seq获得re7292175与RPS26-RTL21共表达的相关性(c图)显著高于bulk RNA从而获得的数据(d图)[4]
——哇哦,那到底如何进行单细胞eQTL分析呀?
——你再往下看,我们都整理好了,惊不惊喜!
单细胞eQTL实验和生信分析框架
正如前文所说,跨不同细胞类型、发育阶段和刺激的eQTL定位创造了巨大的机会,这些在使用bulk RNA测序方法时可能被掩盖。通过降低bulk RNA图谱中由于细胞的异质性而引入的测量噪声,可以识别与罕见细胞类型和特定细胞状态(包括发育阶段、刺激响应状态和细胞周期阶段)相关的eQTL。除了能够区分细胞类型特异的转录组特征外,scRNA-seq研究在识别基因调控网络、基因-基因相互作用和共调控遗传特征方面显示出巨大的潜力。
基于新格元单细胞多组学平台,我们搭建了单细胞eQTL实验流程(图7)和相应分析框架(图8),帮助您挖掘突变-基因-疾病表型三者间的相互作用关系,为疾病机制研究、临床药物研究开发提供强大后援。

图7 单细胞eQTL研究思路框架
有了完备的单细胞eQTL研究框架,还需要有辅之以强的大数据挖掘工具,才能在单细胞大数据时代“入宝山,满载归”。新格元生信与数据中心基于大规模单细胞eQTL研究开发了由浅入深的生物信息分析管线,结合单细胞和WGS的数据特点,充分挖掘单细胞eQTL数据。
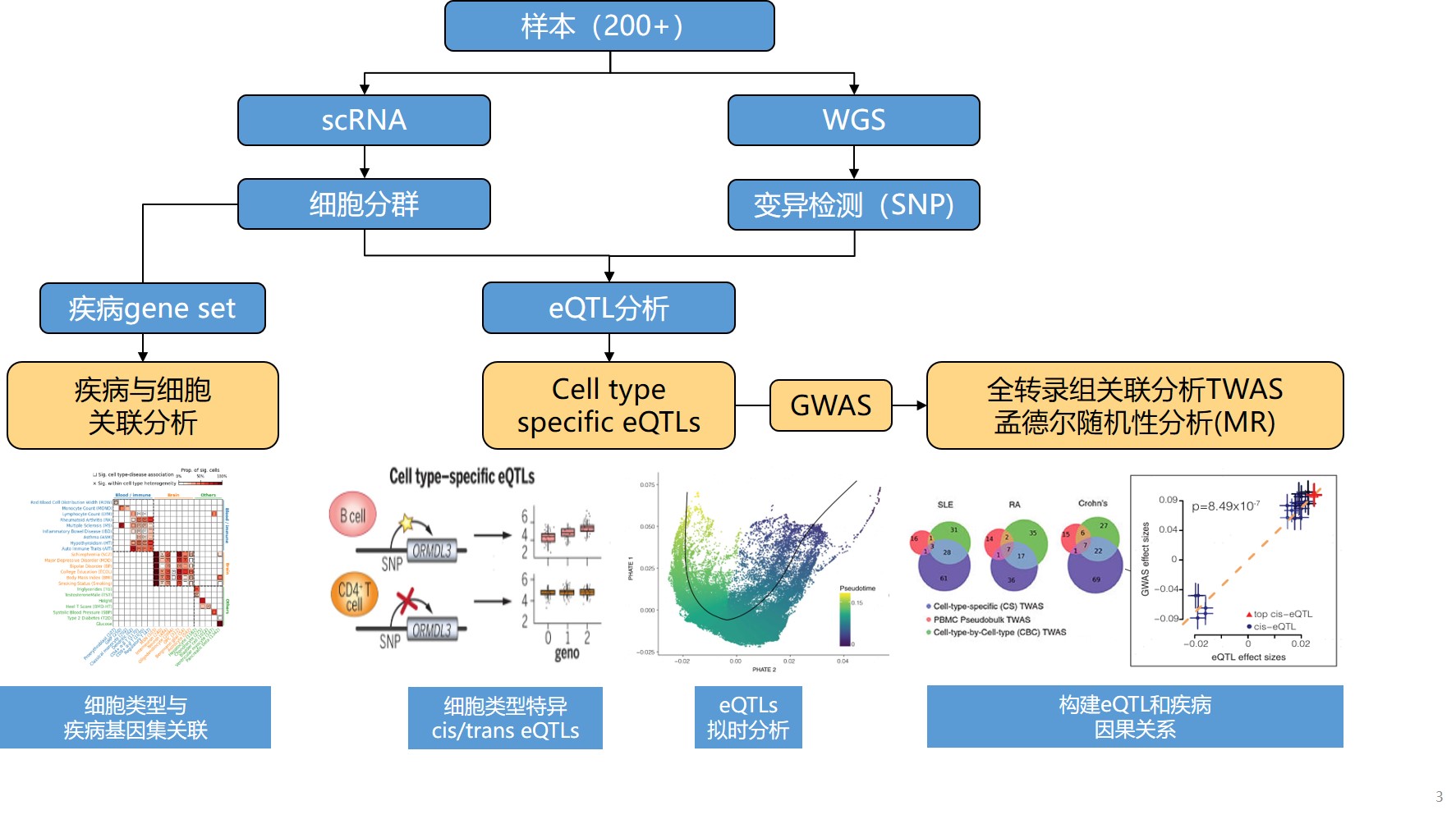
图8 单细胞eQTL生物信息管线
参考文献
[1]Maria M et al., The Power of Single-Cell RNA Sequencing in eQTL Discovery. Genes (Basel) . 2022 Mar 12;13(3):502.
[2]Wills, Q.F.; et al., Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat. Biotechnol. 2013, 31, 748-752.
[3]Kang, H.M.; et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018, 36, 89.
[4]Van der Wijst, et al., Single-cell RNA sequencing identifies cell type-specific cis-eQTLs and co-expression QTLs. Nat. Genet. 2018, 50, 493.
[5]Zhernakova, D.V.; et al. Identification of context-dependent expression quantitative trait loci in whole blood. Nat. Genet. 2016, 49, 139-145.
[6]Zhernakova, D.V.; et al. DeepSAGE Reveals Genetic Variants Associated with Alternative Polyadenylation and Expression of Coding and Non-coding Transcripts. PLoS Genet. 2013, 9, e1003594.







