CellTrek | 将单细胞数据映射到组织切片原位图的精准工具
发布时间:2022-10-21 14:06:30
得益于单细胞测序技术的飞速发展,我们得以从基因表达层面探究不同细胞类型在个体生长发育或疾病发生的动态变化。然而由于缺少细胞在组织中的位置信息,阻碍了细胞微环境的相关研究。新兴的空间转录组测序技术不仅保留了转录组的信息,且能有效保留细胞在组织的空间位置信息。虽然Spatial Transcriptomics、Slide-seq等技术的发展在着力提高分辨率,但目前已商业化的空间转录组技术仍无法达到单细胞水平,只能以混有多个细胞的spot为单位进行研究。如何从spot混杂的细胞中分辨出单个细胞的细胞类型是目前空间技术美中不足之处。
现在普遍使用反卷积的方法,基于特征基因在每个spot的表达占比来拆解出每个spot中的细胞类型。但对一个混合多种细胞类型的spot,反卷积算法难以对其定义标签,而且反卷积算法普通对精细的细胞亚群不敏感。BayesSpace有比反卷积更优的聚类算法,并且不需要单细胞转录组数据的支持。BayesSpace通过拆分spot的方法提高分辨率,并且基于相邻细胞很可能具有相似的转录组特征这一思想,通过构建马尔科夫链模型推测拆分后每个子spot的细胞类型[1]。虽然BayesSpace的表现良好,但其分辨率提升有限且难以结合精准的单细胞转录组注释信息。
为此,新格元引入了新的空间转录组分析流程CellTrek,用于空间转录组数据(ST)和单细胞转录组数据(scRNA-seq)的整合[2]。
1.CellTrek首先将ST和scRNA-seq的原始矩阵合并,接着将合并后的矩阵进行共降维。基于这一降维结果,提取其中的空间转录组数据建立多变量随机森林模型(RF)。
2.随后使用非线性插值的算法提高空间转录组矩阵的分辨率,为更庞大的单细胞转录组数据提供更精细的空间坐标信息。
3.最后将单细胞转录组数据套入RF模型中,构建Spot-Cell表达相似性矩阵为每个单细胞结果添加空间坐标信息并实现可视化作图(图1)。
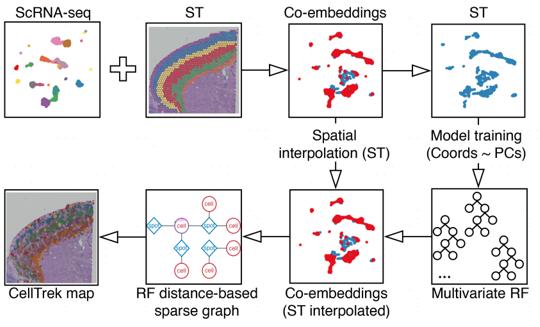
图1 CellTrek主要算法流程[2]
相较于传统拆解spot的做法,CellTrek流程充分利用单细胞转录组数据,直接将单个细胞投影到它们在组织切片原位图中的空间坐标,可以更准确地查看单细胞水平空间位置分布信息,进而开展细胞发育以及细胞互作等微环境的研究。
使用乳腺癌淋巴结数据,对Seurat、BayesSpace和CellTrek提高分辨率后的结果进行了比对(图2)。与基于spot展示的可视化结果相比(图2a),三者都可以体现更高分辨率下每个Spot中可能的细胞类型,且整体细胞类型分布趋势一致。而Seurat结果较为弥散,空间分群边界锐化程度较低,而且有一部分spot的结果比较混杂(图2b)。BayesSpace虽然摆脱了单细胞转录组数据的限制,但其结果固定的将每个Spot均分为6个subspot(10X Visium平台测序数据)无法调节(图2c)。与这两者相比,CellTrek的结果更贴合实际,而且保留了更精准的单细胞转录组细胞注释信息(图2d)。同时CellTrek还支持较为自由的分辨率调节,如改变每个spot所接受的最大细胞数、scRNA-seq数据中单个细胞最多映射到的spot数量等。

图2 不同算法提高分辨率后的结果
由于CellTrek可以保留scRNA-seq细胞注释的结果,我们便将注释信息加入到高分辨率的结果矩阵中,与InferCNV以及拟时序分析进行了接轨(图3,4)。
我们继续使用乳腺癌淋巴结数据,对注释出的肿瘤细胞进行了InferCNV分析,并进行了克隆型划分(图3A)。与CellTrek结果结合,可以清晰地看到不同克隆型的肿瘤细胞具有明显的空间分布差异(图3B)。这为研究肿瘤细胞微环境提供了很好的帮助。
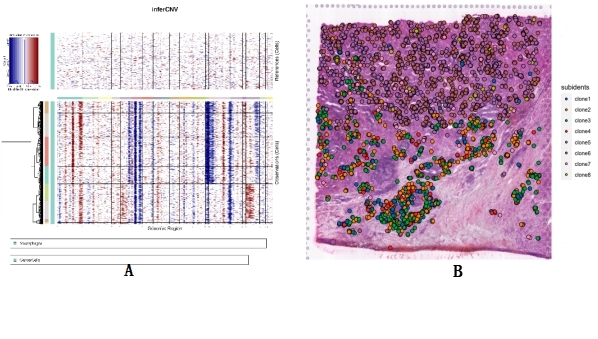
图3 CellTrek与InferCNV结果结合
接下来我们依然使用乳腺癌淋巴结数据,对注释出的巨噬细胞所有亚群进行了monocle2拟时序分析。我们将拟时序生成的细胞注释信息与CellTrek相结合并把结果投射到组织切片上(图4)。CellTrek也很清晰地展示出了巨噬细胞亚群各个细胞在组织切片原位图上的分布情况(图4a)。同时,与拟时序相结合的分析,也将各个细胞pseudotime以及state信息呈现在了组织切片原位图上,呈现了各个细胞分化状态结果在组织切片原位图上的空间分布情况(图4b,c)。
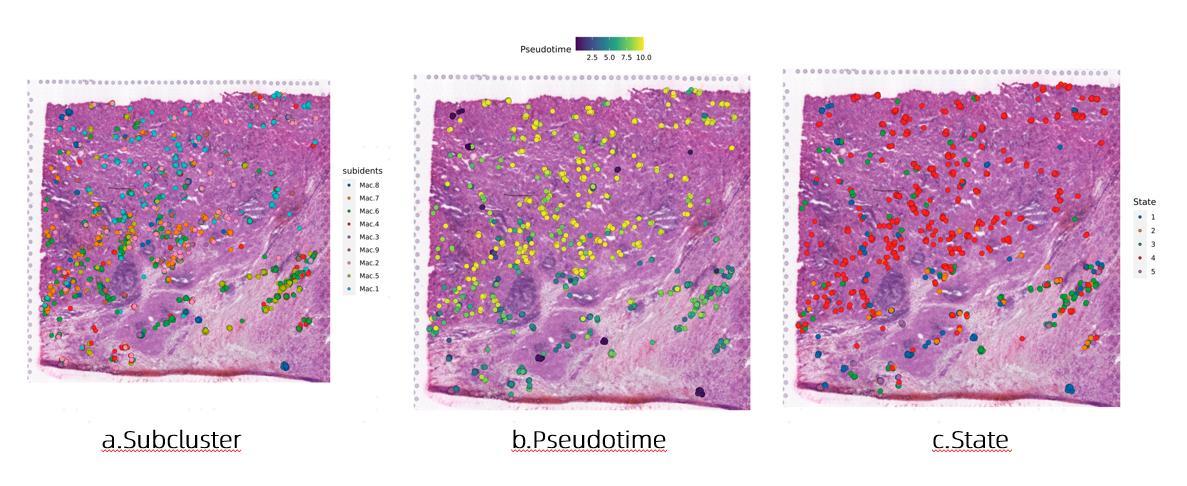
图4 CellTrek与亚群细分以及拟时序分析的结合
得到了不同细胞类型在组织切片上的分布后,CellTrek还提供了SColoc和SCoexp两个下游模块,分别来分析不同细胞类型之间的空间关联性以及不同表达模式的module在空间位置分布情况。
SColoc模块可以计算各个细胞类型之间的空间相似性矩阵,基于空间相似性矩阵构建最小生成树代表细胞类型间的空间关系。图5a为SColoc模块基本算法流程。图5b为各个细胞类型空间共定位的结果。圆圈大小表示细胞类型数量,连线粗细表示空间位置权重大小,连线长短无实际意义。预测结果显示肿瘤细胞与巨噬细胞有更强的关联性,结合图2看两者在空间分布上确实更加靠近。但两者是否有互作关系还需要进一步的分析验证。
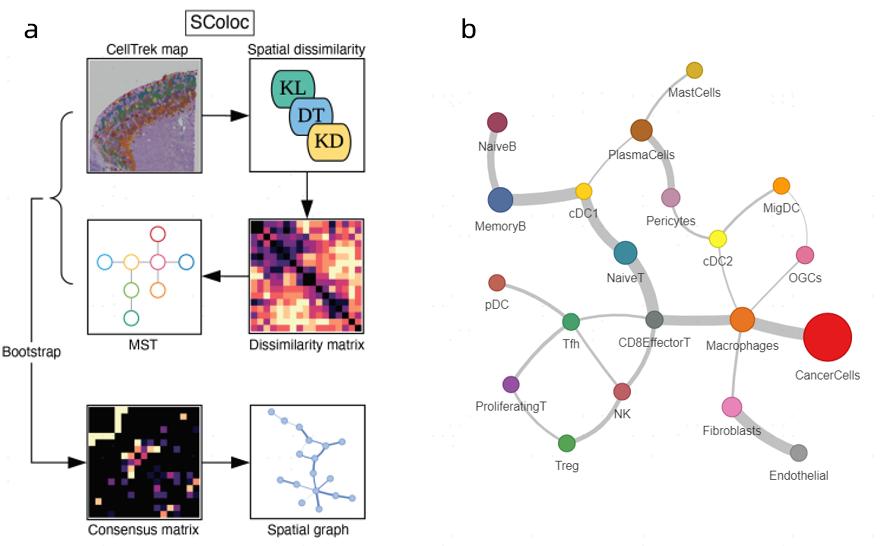
图5 SColoc模块分析不同细胞类型空间关联性
SCoexp根据空间距离构建spatial-weight基因共表达矩阵,然后通过Contrastive Clustering或者WGCNA聚类将共表达基因划分为不同的Module。目前使用Seurat的AddModuleScore打分,并输出各个Module得分可视化结果。图a为SCoexp模块基本算法流程,图b、c分别展示了不同module的得分在组织切片原位图以及降维图中的分布情况。结果显示通过共表达基因鉴定出了3个模块,而肿瘤相关module(module1)和免疫相关module(module2,module3)在空间分布上具有明显的差异。
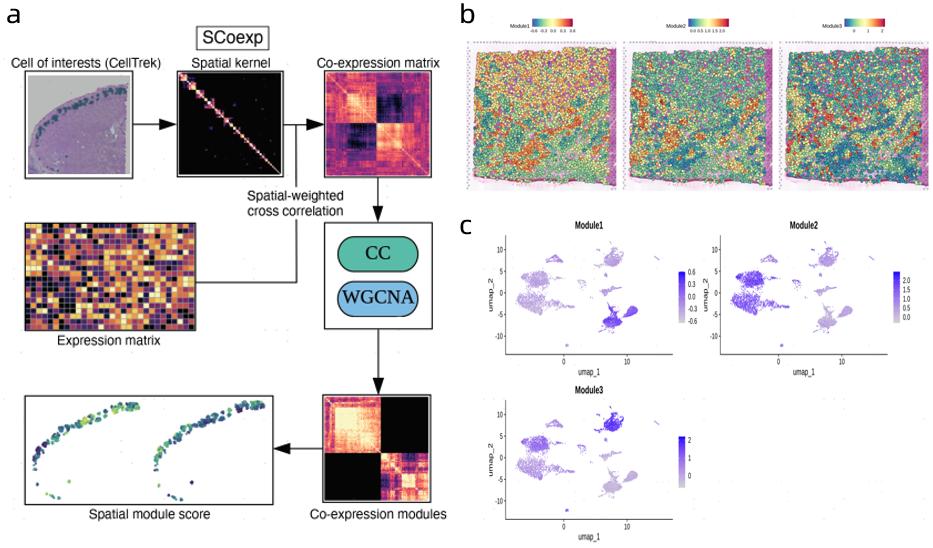
图6 SCoexp模块共表达基因集合分析
总之CellTrek可利用ST的数据,为scRNA-seq的每个细胞预测空间分布位置,可以有效地帮助研究者深度挖掘组织微环境的生物信息。
现在,CellTrek流程已经正式纳入新格元空间转录组分析板块,快带上数据前来探索吧!
参考文献
[1] Zhao, E., Stone, M.R., Ren, X. et al. “Spatial transcriptomics at subspot resolution with BayesSpace.” Nat Biotechnol 39, 1375–1384 (2021).
[2] Wei R, He S, Bai S, Sei E, Hu M, Thompson A, Chen K, Krishnamurthy S, Navin NE. Spatial charting of single-cell transcriptomes in tissues. Nat Biotechnol. 2022 Aug;40(8):1190-1199. doi: 10.1038/s41587-022-01233-1. Epub 2022 Mar 21. PMID: 35314812.







