如虎添翼丨新格元scRNA联合WES分析助力肿瘤CNV研究
发布时间:2022-10-21 14:46:12
单细胞转录组测序(single cell RNA Sequencing,scRNA)和外显子组测序(Whole Exon Sequencing,WES)分析广泛应用于人类肿瘤疾病研究。两者都可以通过分析测序数据的基因拷贝数变异(Copy number variations,CNV)情况从而助力肿瘤研究。通过对比scRNA和WES在CNV分析中的优劣势(见表1),可以看到,scRNA可以很好的获得不同细胞的CNV情况。但是scRNA是基于转录组推断的CNV,与真实的基因组CNV相比可能存在一定的误差,并且覆盖度相对比WES要低;而WES是通过捕获DNA序列进行的测序,因此可以相对比较准确获取CNV的情况,但无法获得每个细胞的CNV情况。综上所述,两种技术在CNV分析应用中互有优劣。
为了更深入的探究肿瘤细胞中CNV情况对疾病发展进程的影响,新格元特别推出单细胞联合WES分析。
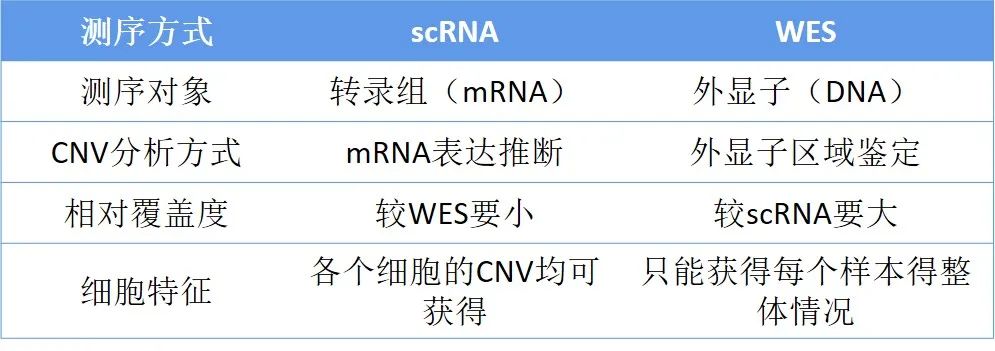
表1 scRNA与WES在CNV分析方面的优劣势
考虑到WES分析的特性[1],获得体细胞突变(Somatic Mutation)需要肿瘤病理组织样本及同一个人配对的正常组织样本(可以是癌旁组织,也可以是血液样本),而单细胞本身可以鉴定出一群正常的细胞,以此正常细胞为参考获得体细胞突变特征。整体实验方案如下图1,WES需要同一个病人的配对样本,scRNA可以仅有肿瘤样本也可以配对样本进行。
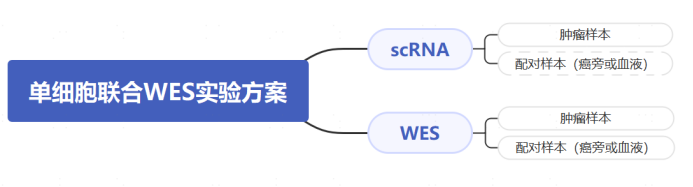
图1 单细胞联合WES实验方案(注:配对样本指肿瘤样本的相同个体的正常癌旁或血样样本)
单细胞联合WES分析方案如下图所示(图2)。首先分别对scRNA数据和WES数据分别进行CNV分析,基于上述结果进行联合分析。联合分析内容主要包括:
3. 共有扩增/缺失染色体区段基因富集分析。
通过两者联合分析能够更加准确的定位变异部位,为CNV数据挖掘提供一定的方向。
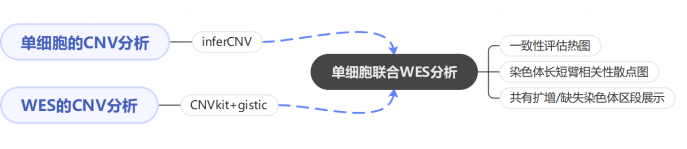
图2 单细胞联合WES分析方案
对于单细胞CNV分析,我们最常用的分析工具为InferCNV[2]。该软件用于推断scRNA水平的基因拷贝数变异情况,比如整条染色体或大片段染色体的扩增或缺失,可被应用于恶性肿瘤细胞识别,肿瘤异质性分析等方面。图3是inferCNV结果按样本的信息进行细胞重新排序展示的热图。可以看到不同样本表现出不同的CNV克隆异质性。
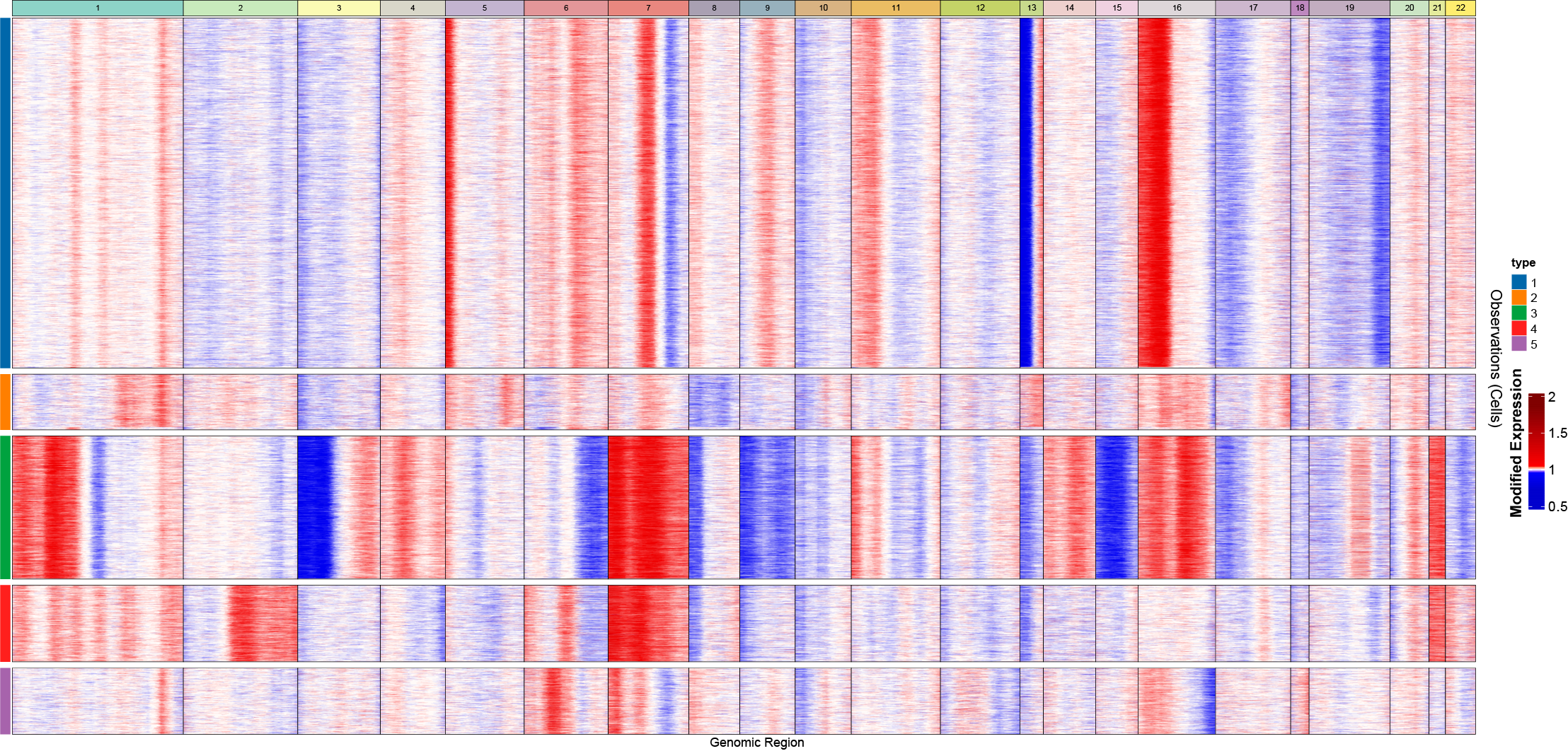
图3 以样本信息重新排序展示inferCNV热图(红色代表CNV扩增,蓝色代表CNV缺失)
CNVkit[3]和gistic[4]工具用于WES的CNV分析。CNVkit是用来检测全基因组和外显子组CNV的工具,该工具主要适用于多个配对样本测序情况,首先对所有的normal数据进行bin处理拿到背景值,然后就这个背景值来处理所有的case测序数据计算拷贝数变异情况。CNV分析中的体细胞CNV鉴定,不仅仅需要病理组织,还需要配对的同一个个体的正常组织(可以是正常血液样本)进行对照配对,才能获得比较准确的体细胞CNV情况,便于从中获取同研究实验因素条件下相关的CNV。基于CNVkit分析结果,利用gistic软件分析各个CNV显著扩增或缺失的基因组区域,进而获得每个基因的CNV情况。图4展示了gistic分析后的各个样本WES数据中的CNV情况,可以与scRNA的inferCNV进行组合对照展示。
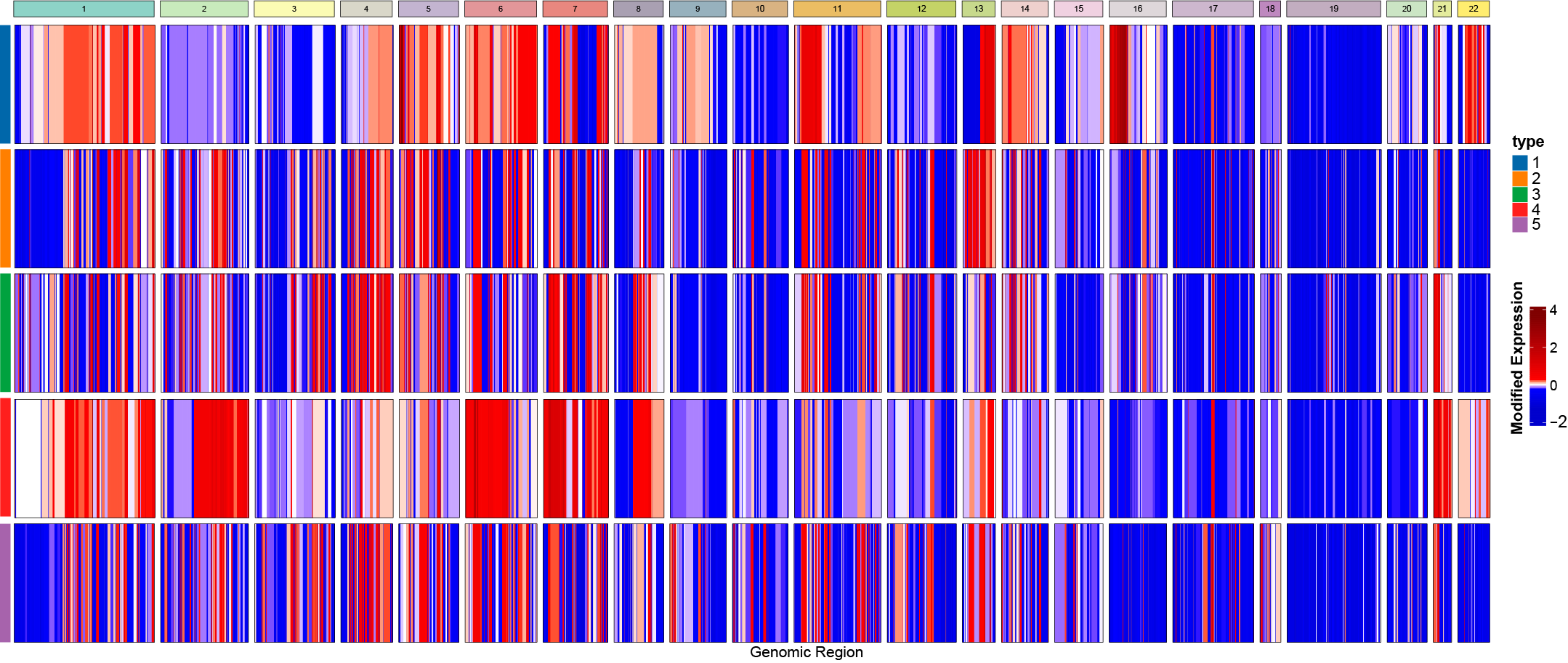
图4 WES数据CNVkit+gistic分析后热图(红色代表CNV扩增,蓝色代表CNV缺失,每行代表一个样本)
为了更准确地定位发生CNV变异的区域,首先对scRNA和WES分析结果进行一致性评估[5]。由于每个个体的CNV情况不一致,这里推荐基于单样本分析结果进行一致性评估,进而获得每个样本中CNV情况相对一致的染色体区段。图5展示了各个样本的scRNA与WES的CNV一致性评估热图结果,可以看到红色和白色部分比较多的样本,其scRNA与WES的结果一致性程度更高,如1号样本中16号染色体和19号染色体存在部分染色体扩增区段在scRNA与WES的结果是相对一致的,这些一致的CNV事件发生可能与该疾病相关,可以针对该CNV区段事件进行进一步探索分析。
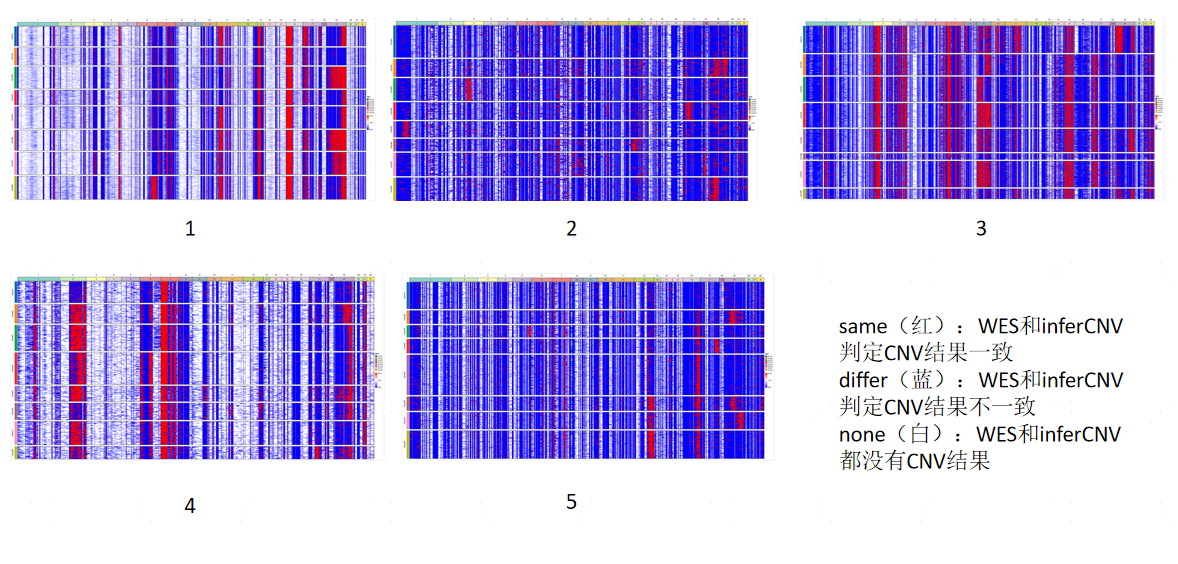
图5 scRNA与WES的CNV一致性评估热图(红色为WES和scRNA的相同CNV事件,蓝色为WES和scRNA的不同CNV事件,白色为WES和scRNA均没有鉴定出CNV事件)
为了更好评估scRNA与WES的结果一致性,我们还可以对两种数据中CNV事件在染色体长短臂中的表现进行相关性分析[6],如下图6就展示了1号样本的情况,可以看出1号样本在scRNA与WES中CNV的长短臂表现出很高的相关性,结合热图可以更好反映了该样本CNV事件在两类数据的一致性情况。
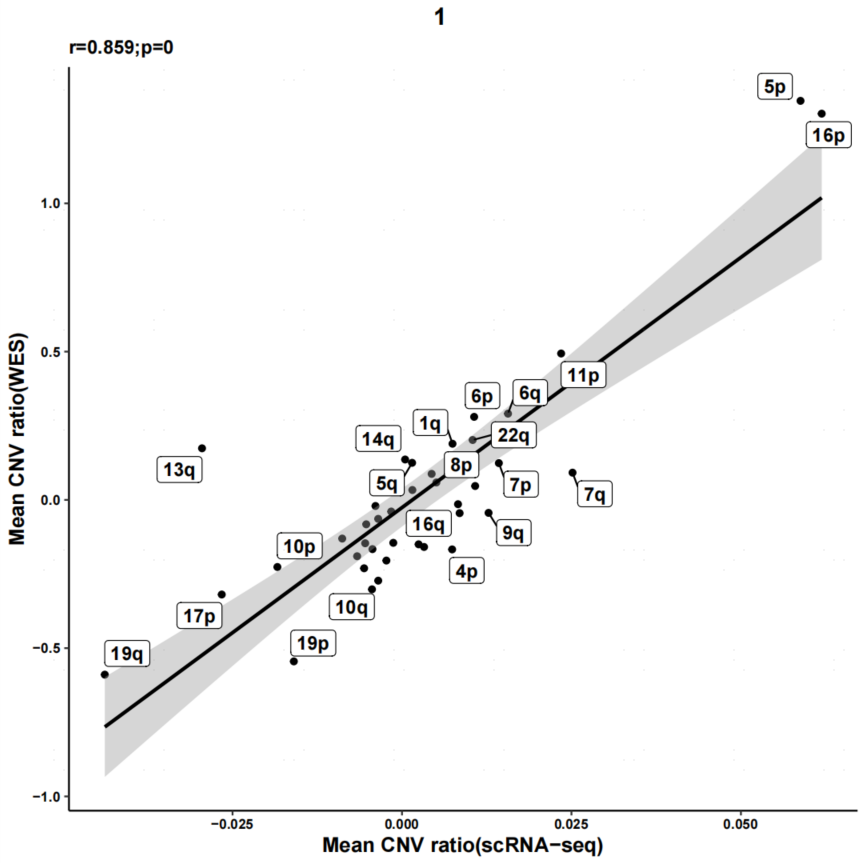
图6 scRNA与WES的CNV长短臂相关性散点图
除一致性评估以外,还可以分别对scRNA数据与WES数据进行染色体区段展示,图7a和b分别展示了1号样本染色体区段在scRNA数据和WES数据中的扩增缺失情况。如图所示16号染色体部分区段的在scRNA与WES数据中保持一致的扩增趋势,19号染色体部分区段在scRNA与WES数据中保持一致的缺失趋势。
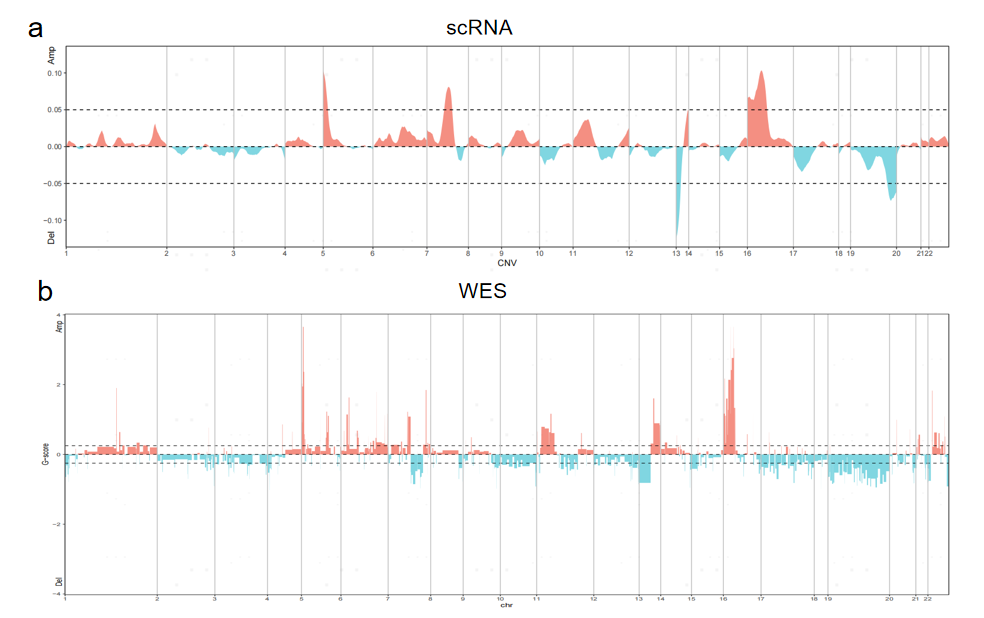
图7 scRNA数据和WES数据染色体区段展示CNV事件情况(a:scRNA数据;b:WES数据)
除从基因层面揭示DNA扩增引起表达提高的现象外,还可以通过分析scRNA与WES结果的基因列表的交集及其对应的染色体位置,反映CNV在scRNA与WES数据的共性表现情况。通过分析scRNA和WES数据中共有的某个染色体区段的结果可以对该共有部分的基因进行功能富集,从功能上探究该染色体相关事件可能对研究对象的影响。图8展示了1号样本scRNA数据与WES数据中16号染色体扩增的基因交集情况Venn图。最终可以通过结合这些基因在正常组织和疾病组织之间的是否存在表达差异,寻找到一群在CNV事件中与疾病发生发展相关的基因。
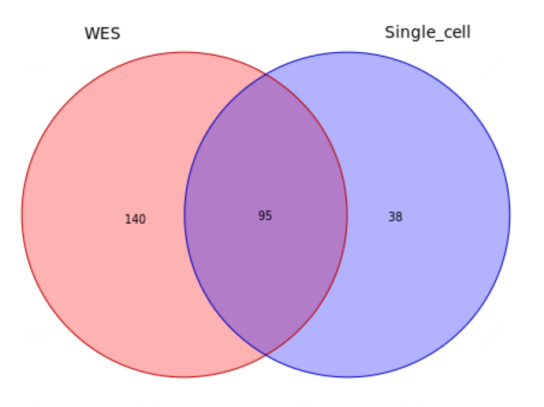
图8 scRNA数据与WES数据中16号染色体扩增基因交集Venn图
目前整体方案各取scRNA数据与WES数据之优势,通过多组学手段联合,珠联璧合,相互印证,多维度、多角度进行分析,使得分析更加准确和可靠,希望能对肿瘤研究在CNV层面提供一种新的分析思路和方法,最终加速肿瘤的研究进程。
以上就是新格元scRNA和WES联合分析方案,后续我们会根据不同的情况及分析目的继续进行优化,以期提供更加全面及优质的生物信息数据挖掘服务。
参考文献
[1]Koboldt Daniel C,Best practices for variant calling in clinical sequencing.[J] .Genome Med, 2020, 12: 91.
[2]Patel Anoop P,Tirosh Itay,Trombetta John J et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma.[J] .Science, 2014, 344: 1396-401.
[3]Talevich Eric,Shain A Hunter,Botton Thomas et al. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing.[J] .PLoS Comput Biol, 2016, 12: e1004873.
[4]Mermel Craig H,Schumacher Steven E,Hill Barbara et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers.[J] .Genome Biol, 2011, 12: R41.
[5]Sun Yunfan,Wu Liang,Zhong Yu et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma.[J] .Cell, 2021, 184: 404-421.e16.
[6]Wang Ruiping,Dang Minghao,Harada Kazuto et al. Single-cell dissection of intratumoral heterogeneity and lineage diversity in metastatic gastric adenocarcinoma.[J] .Nat Med, 2021, 27: 141-151.







