新品发布 | 单细胞转录组二代+三代测序解决方案
发布时间:2022-11-30 14:47:47
单细胞转录组测序(scRNA-seq)技术作为研究复杂生物体的利器,能够揭示单个细胞的分子状态,解析组织异质性。主流的单细胞转录组基于短读长二代测序策略直接研究细胞类型特异的可变剪切,融合基因,isform等还稍欠火候。
在单细胞全长转录组测序中,我们介绍了三代单细胞转录组的相关内容。相较于单独的二代测序,三代测序主要有两大优势:
1.测序过程中不需要PCR扩增,避免PCR偏好性;
2.长读长,可以对转录本全长进行测序。
将二代测序与三代测序相结合,能够在成本适中的前提下,既获得转录本的全长序列,又能够通过Cell Barcode信息定位到细胞亚群(图1)。
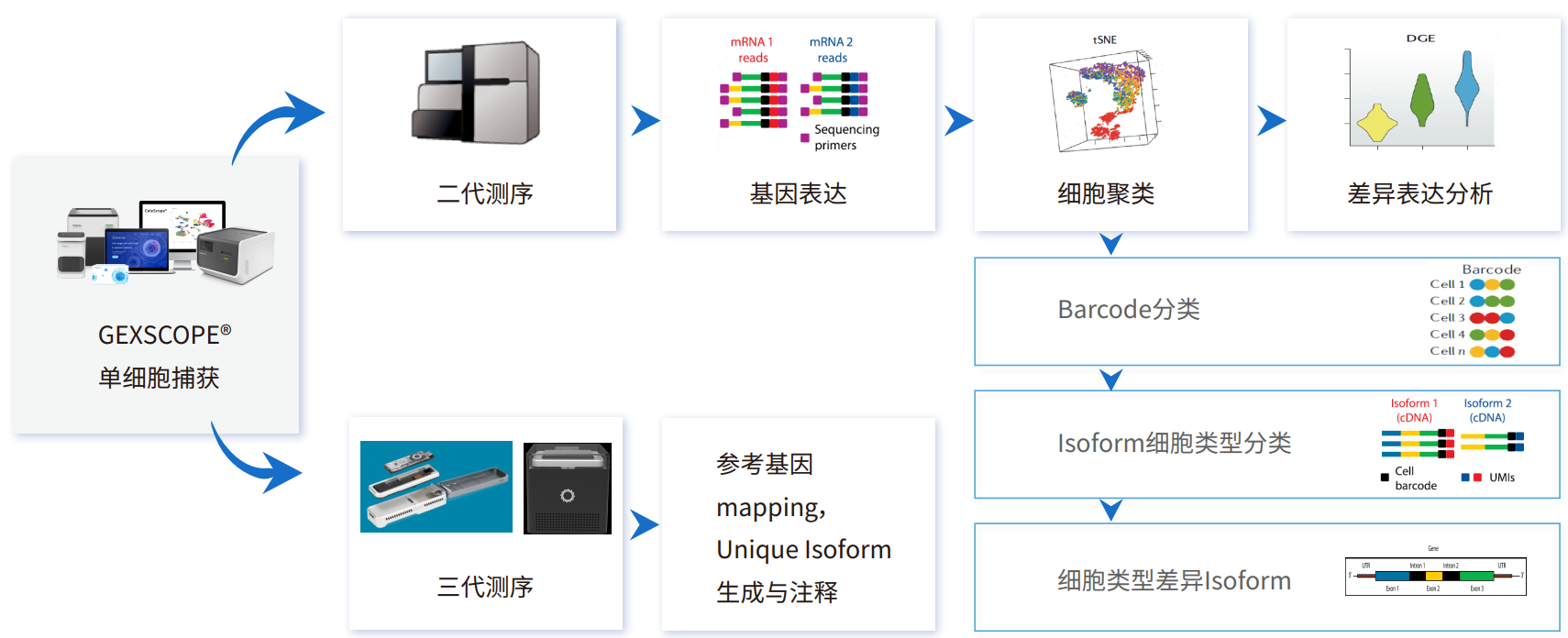
图1 单细胞转录组二代+三代测序策略
在单细胞二代测序的基础上,新格元推出单细胞转录组二代+三代测序方案。新增全长检测的三代测序,分析内容包括SNP/CNV、基因可变剪切、TCR/BCR全长检测、转录本isoform等。能够在单细胞水平上研究关键基因的转录本并发现新的转录本或关键基因的新的可变剪切类型,在遗传发育、肿瘤发生等方向有着重要应用。
新格元单细胞转录组二代+三代解决方案,利用GEXSCOPE®平台对单细胞mRNA捕获后进行反转录生成cDNA,利用同一份cDNA分成两部分,分别构建二代测序文库和三代测序文库,流程优化简单,二、三代单细胞优势互补,大幅降低整体实验成本,为单细胞全长转录组测序提供了一站式解决方案。并且在数据分析上相较于单独的单细胞二代或三代测序,单细胞二代+三代能够得到更加精细的细胞聚类和细胞注释。三代测序的超长读长,可获得mRNA全长序列及结构信息。
单细胞转录组二代+三代测序的潜在应用范围广泛,目前已成功应用于识别细胞周期、肿瘤细胞异质性研究、鉴定罕见细胞类型、免疫细胞分型、干细胞分型、疾病分型等领域。更多方向详见图2。
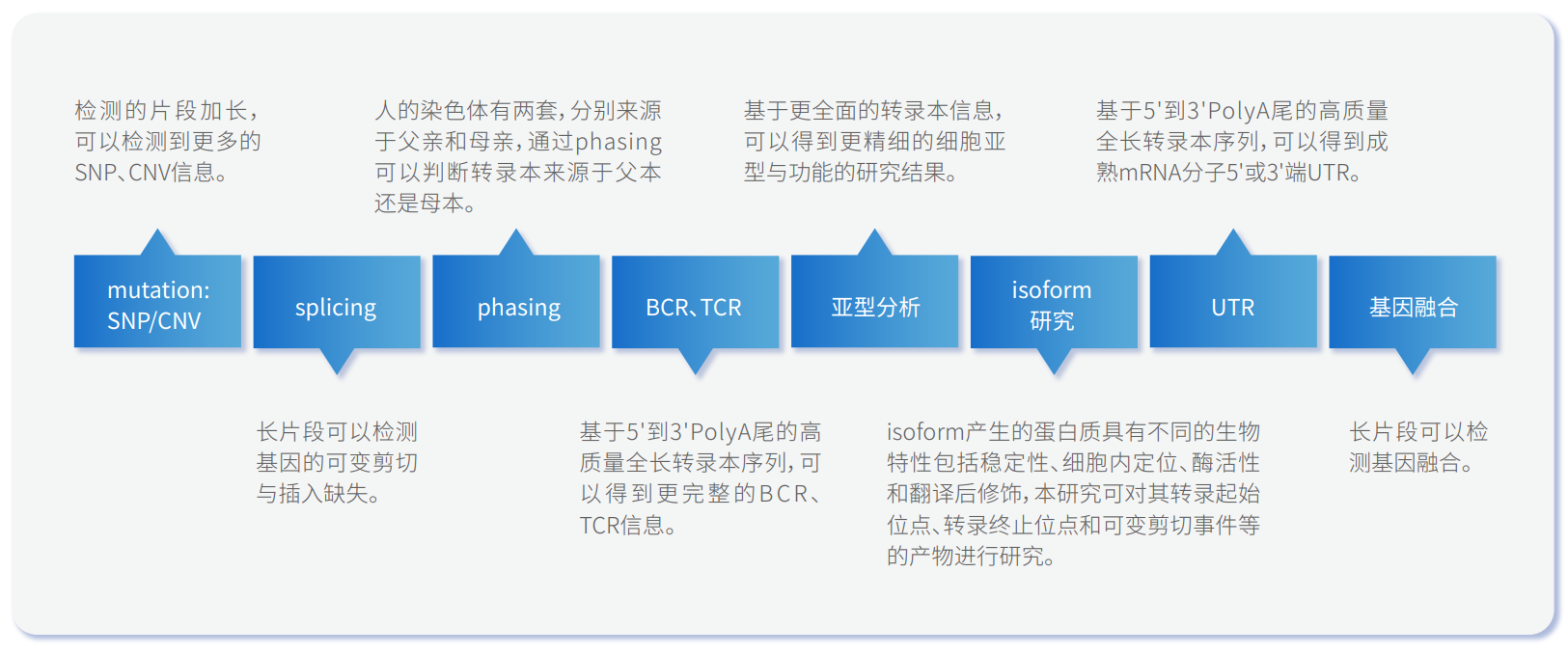
图2 单细胞转录组二代+三代测序联合应用
文章主题:Transcriptome variation in human tissues revealed by long-read sequencing
发表杂志:Nature
发表时间:2022年8月
前体mRNA的选择性剪切可在同样的DNA序列中产生不同的蛋白质,在细胞分化以及个体发育中起着重要的作用。通常认为,在大多数情况下,一个基因通常注释2个转录本。而下面这篇研究在已注释基因中发现了更多的新转录本。
在这篇今年8月份的Nature文章中1,研究人员利用三代纳米孔测序,对来自GTEx(Genotype-Tissue Expression)56个供体和4个K562细胞系总共90个样本进行了测序,并同时使用二代测序和质谱进行验证,这是迄今为止最大的长reads RNA数据集研究。通过对转录本进行定量分析并鉴定新的转录本,结果在21067个基因中发现了93718个转录本,其中77%是新发现的转录本。
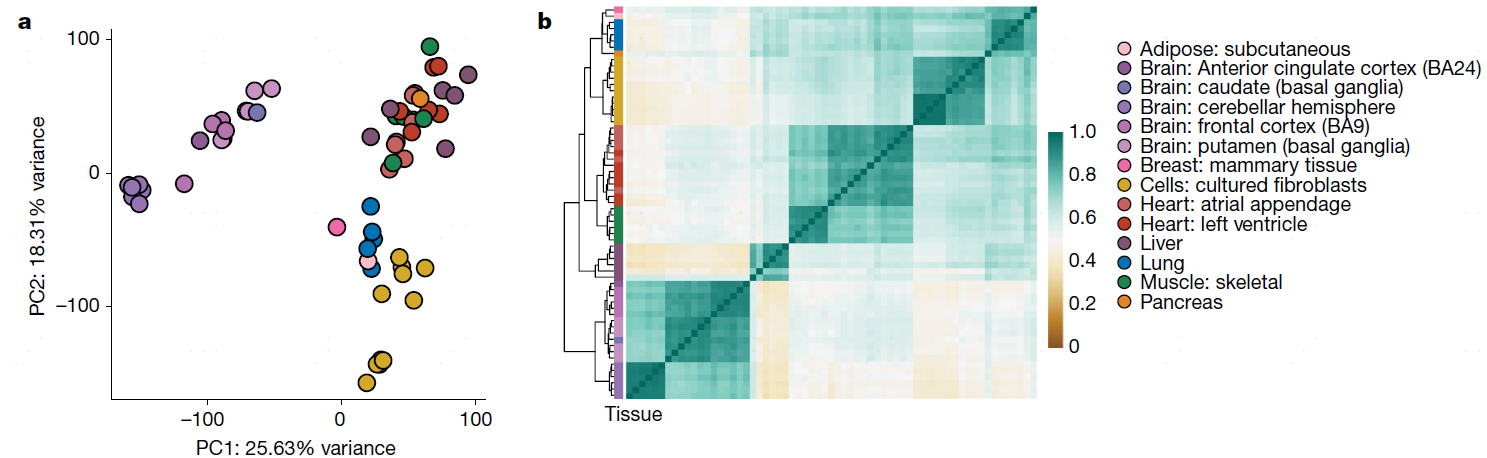
图3 新转录本isoform的发现
文章主题:Comprehensive characterization of single-cell full-length isoforms in human and mouse with long-read sequencing
发表杂志:Genome Biology
发表时间:2021年11月
等位基因特异性表达(ASE)是另一个值得关注的问题。在特定的个体中,基因组的两个等位基因拷贝,不一定都是活跃的或在同一水平上,这种不平衡即ASE。在肿瘤细胞以及性染色体基因中,这种现象尤为明显。
在2021年发表在Genome biology上的一篇文章中2,研究人员对人类肿瘤细胞和小鼠肌肉干细胞进行单细胞全长测序,以实现单细胞中的异构体发现、剪接分析和突变检测。研究人员确定了数千种未注释的isoforms,并发现了保守的功能模块。通过聚类分析发现,人类肿瘤细胞(CLL)相比正常免疫细胞具有更高比例的新型转录本,尤其是新型剪接的转录本。通过对这些变异的等位基因频率进行分析,研究人员确定了两个亚克隆。BCL2、RAD21和PHEX基因中的SNV在clone1中富集,而位于B3GAT3基因中的突变在clone2中富集(图4)。
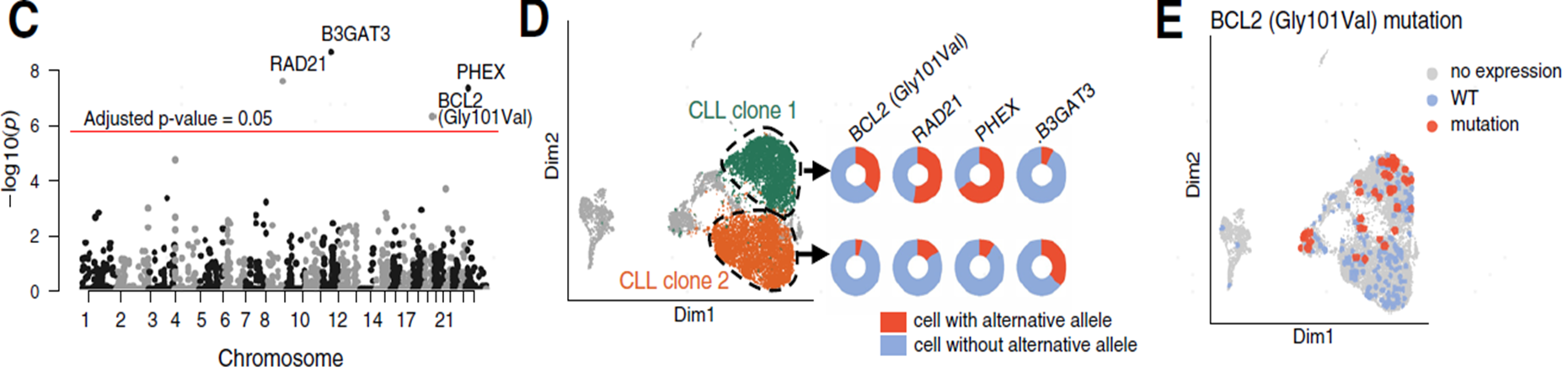
图4 等位基因频率分析
文章主题:JAFFAL: detecting fusion genes with long-read transcriptome sequencing
发表杂志:Genome Biology
发表时间:2022年1月
融合基因是染色体畸变(chromosomal abberations)的结果,由染色体重排产生。融合基因的产生改变了基因的蛋白编码序列或调控序列,使得基因功能发生变化,对机体的影响较大。在某些癌症类型的细胞中有着重要的作用。
在2022年发表在Genome biology上的一篇文章中3,研究人员开发了一种名为JAFFAL的算法,首先对基于三代的肿瘤和健康细胞系数据进行分析,发现在cDNA文库制备过程中产生了大量的嵌合分子,而RNA直接测序中不存在这些嵌合分子。随后,研究人员又利用该算法,对5种肿瘤细胞系的单细胞结果进行分析,结果在H838非小细胞肺癌细胞系中鉴定到了由三个基因组成的融合基因BMPR2-TYW5-ALS2CR11,展示了该算法从复杂重排中检测单细胞融合基因的能力(图5)。
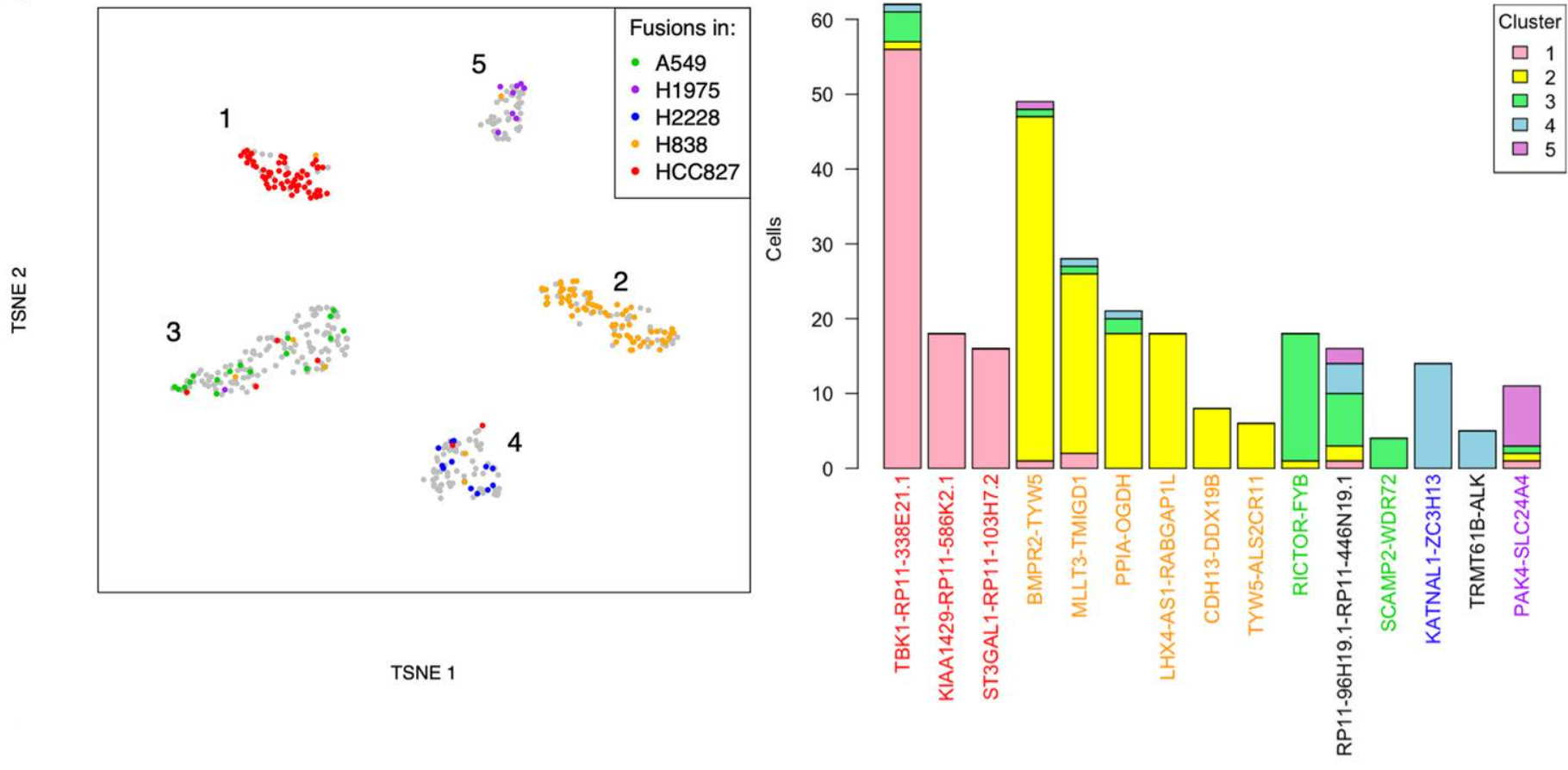
图5 融合基因分析
新格元专注于单细胞多组学全流程产品与服务的开发,目前经过不断的优化研发,能够提供单细胞转录组二代+三代测序分析服务。有想了解的小伙伴可以通过微信后台或当地销售与我们联系哦,速速call我们吧!
1、Glinos DA, Garborcauskas G, et.al. Transcriptome variation in human tissues revealed by long-read sequencing. Nature. 2022 Aug;608(7922):353-359.
2、Tian L, Jabbari JS, Thijssen R, et.al. Comprehensive characterization of single-cell full-length isoforms in human and mouse with long-read sequencing. Genome Biol. 2021 Nov 11;22(1):310.
3、Davidson NM, Chen Y, et.al. JAFFAL: detecting fusion genes with long-read transcriptome sequencing. Genome Biol. 2022 Jan 6;23(1).







