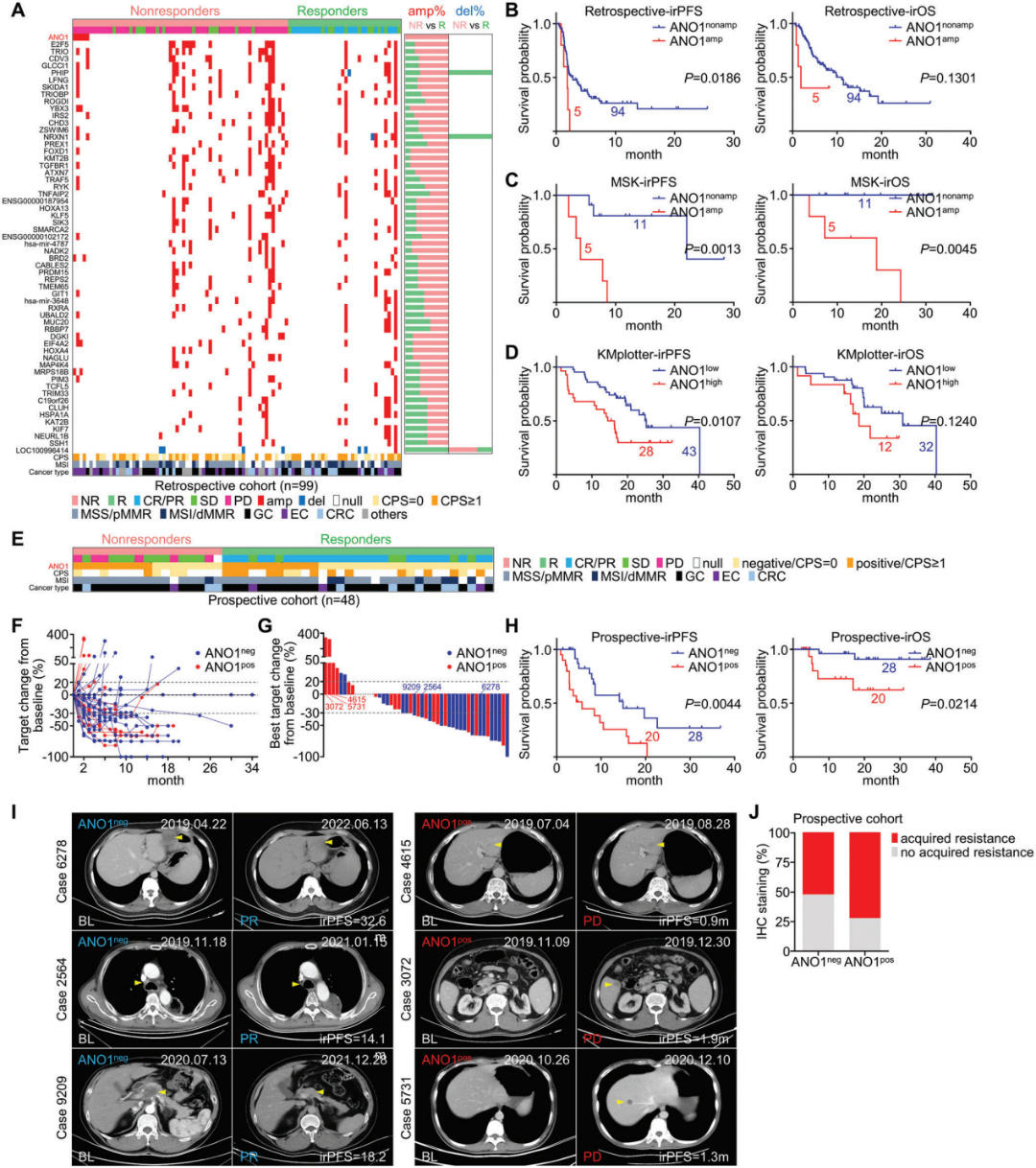
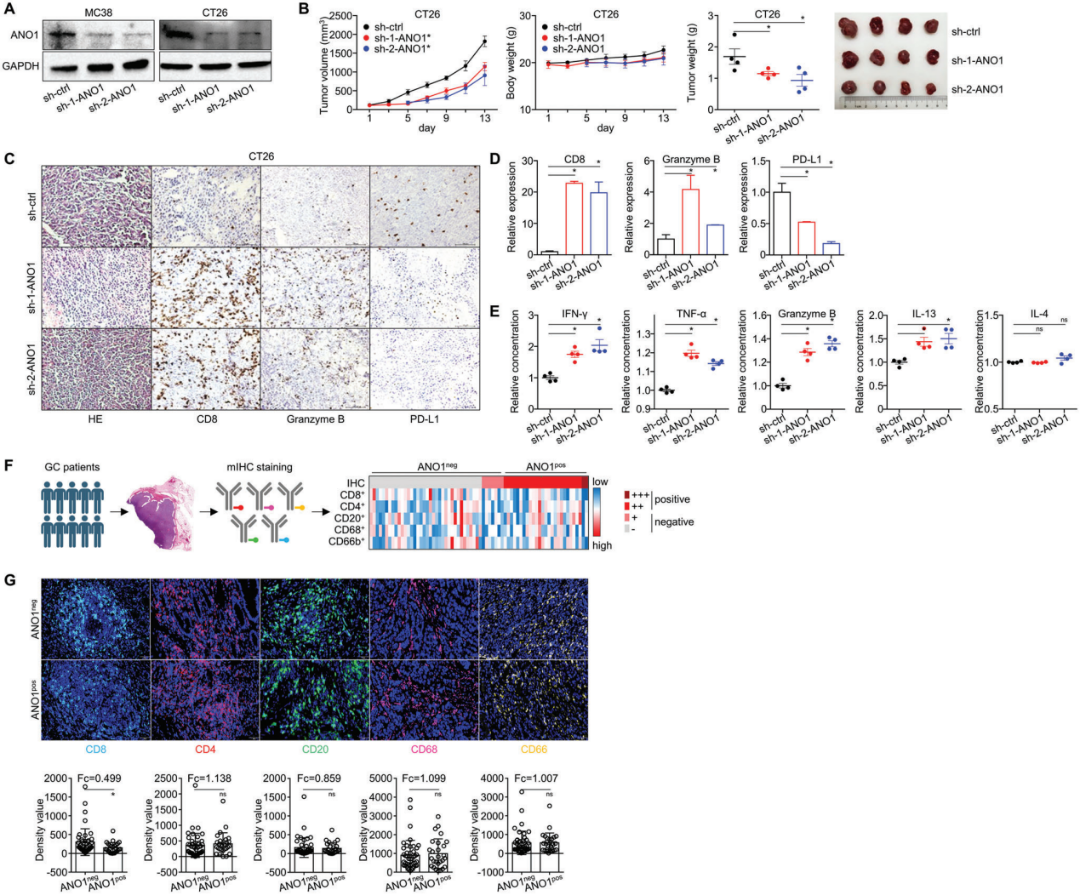
图1. ANO1扩增或高表达可预测免疫治疗不良预后
在转录水平上,ANO1在GC/EC/CRC肿瘤中的表达高于正常组织(图S2B)。在蛋白水平上,70例胃癌手术患者的病灶免疫组化结果(图S2D-F)也显示,肿瘤组织中ANO1的阳性率明显高于正常组织。
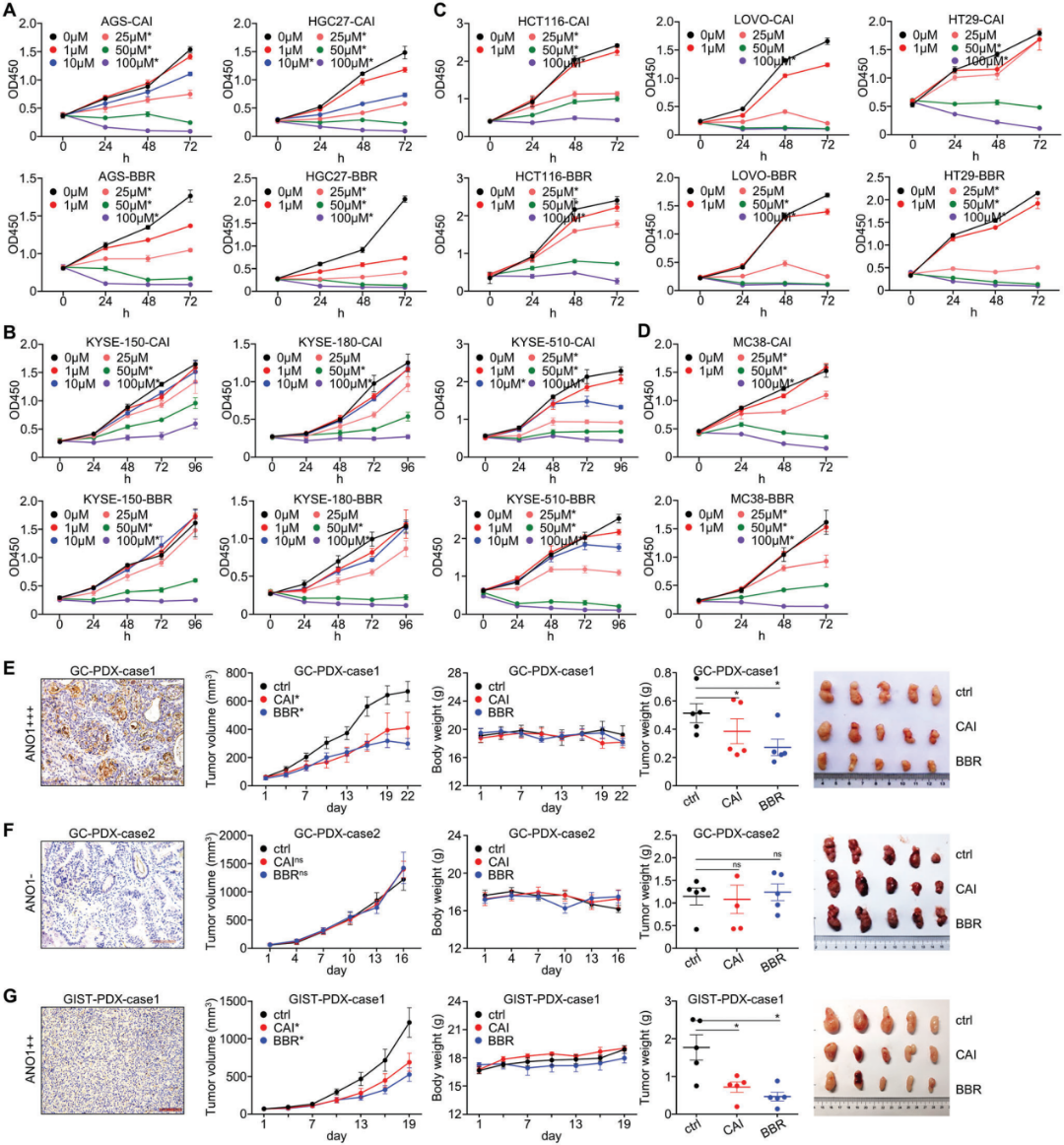
为了进一步验证ANO1作为GI癌症治疗靶点的潜力,作者评估了之前报道的两种针对ANO1的抑制剂CaCCinh-A01(CAI)和苯溴马酮(BBR),在细胞系和病人来源的异种移植(PDX)模型中作用效果。CAI/BBR可抑制PDX模型(图S4A,B),及体外培养GC(图3A)、EC(图3B)、CRC细胞株(图3C)和小鼠癌细胞MC38(图3D)的ANO1蛋白表达。此外,CAI/BBR选择性地在ANO1阳性的GC PDX中(图3E),而不是在ANO1阴性的GC PDX中(图3F),显示出抗肿瘤作用,并且能有效抑制ANO1阳性的GIST PDX模型的生长(图3G)。这些发现支持ANO1是GI癌症靶向治疗的药物靶点。
图3. ANO1是胃肠道癌症靶向治疗的可用药靶点
4.ANO1促进免疫抑制肿瘤微环境
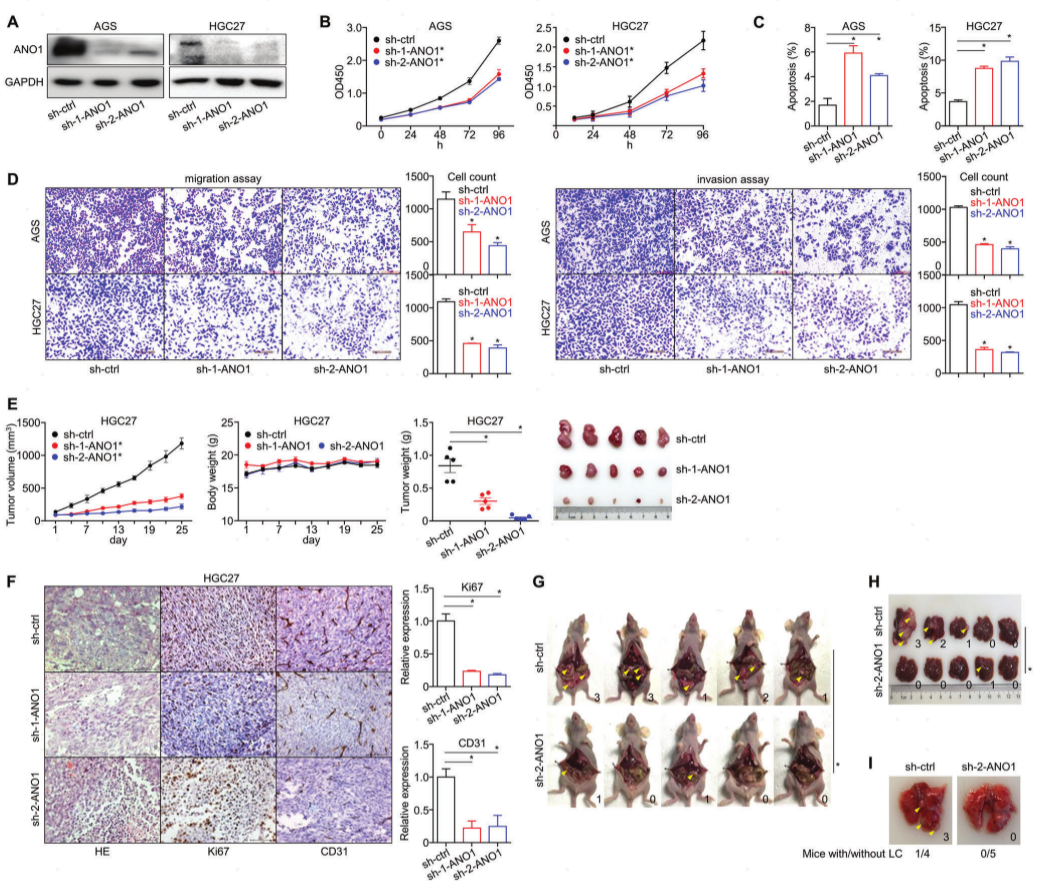
对小鼠结肠癌(COAD)细胞株进行了ANO1敲除(图4A)。与免疫缺陷人肿瘤CDXs的结果相比较,ANO1敲除降低了CT26 CDXs的生长(图4B),同时增加了异种移植肿瘤中CD8/granzyme B的含量,降低了PD-L1表达(图4C,D),提示细胞毒性CD8+ T细胞在TIME期的浸润增加。根据小鼠CDXs的ELISA检测,在异种移植瘤中IFN-γ/granzyme B/TNF-α/IL-13表达显著升高,而免疫抑制细胞因子IL-4在ANO1敲除后基本保持不变(图4E)。作者还对前面提到的70例胃癌手术病例的主要免疫细胞进行了多重IHC(图4F),发现ANO1阳性组的CD8+ T细胞浸润明显低于ANO1阴性组(图4G)。因此,ANO1有助于免疫抑制肿瘤微环境,而敲除ANO1可以及时恢复免疫活性。
图4. ANO1形成免疫抑制肿瘤微环境
5.ANO1抑制增强了抗PD-1免疫治疗对胃肠道肿瘤的有效性
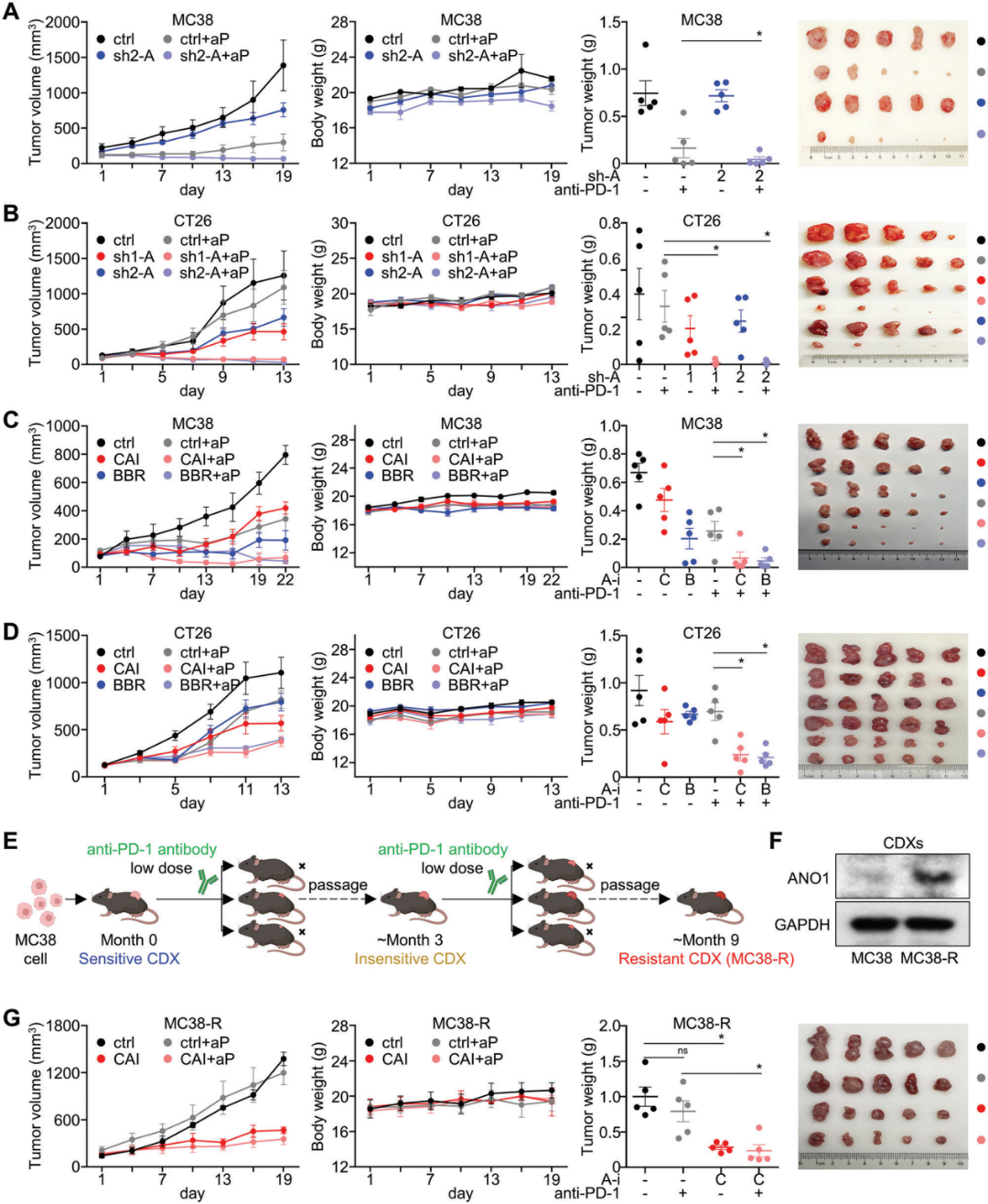
由于ANO1敲低可以破坏免疫抑制的肿瘤微环境,作者评估了抑制ANO1对MC38或CT26移植的C57BL-6J/BALB-C小鼠模型中抗PD-1抗体有效性的影响。MC38异种移植瘤对抗PD-1抗体表现出高敏感性(图5A),而CT26异种移植瘤对抗PD-1抗体表现出低敏感性(图5B)。ANO1敲除(图5A,B)或使用CAI/BBR抑制剂(图5C,D)均可明显增强抗PD-1抗体对MC38/CT26异种移植瘤的抗肿瘤作用。作者还通过抗PD-1抗体对MC38进行长期、低剂量的诱导,建立了MC38- R免疫治疗的获得性耐药(图5E)。作者观察到,与MC38-R CDX的祖细胞MC38 CDX相比,在MC38-R CDX中ANO1的表达升高(图5F),强调了ANO1参与了抗PD-1免疫治疗的获得耐药性。值得注意的是,ANO1抑制剂CAI也使MC38-R CDX的抗PD-1抗体敏感度提升(图5G)。
图5.抑制ANO1可增强抗PD-1免疫治疗对胃肠道癌症的有效性
6.ANO1通过抑制铁死亡促进肿瘤进展并损害免疫治疗效果
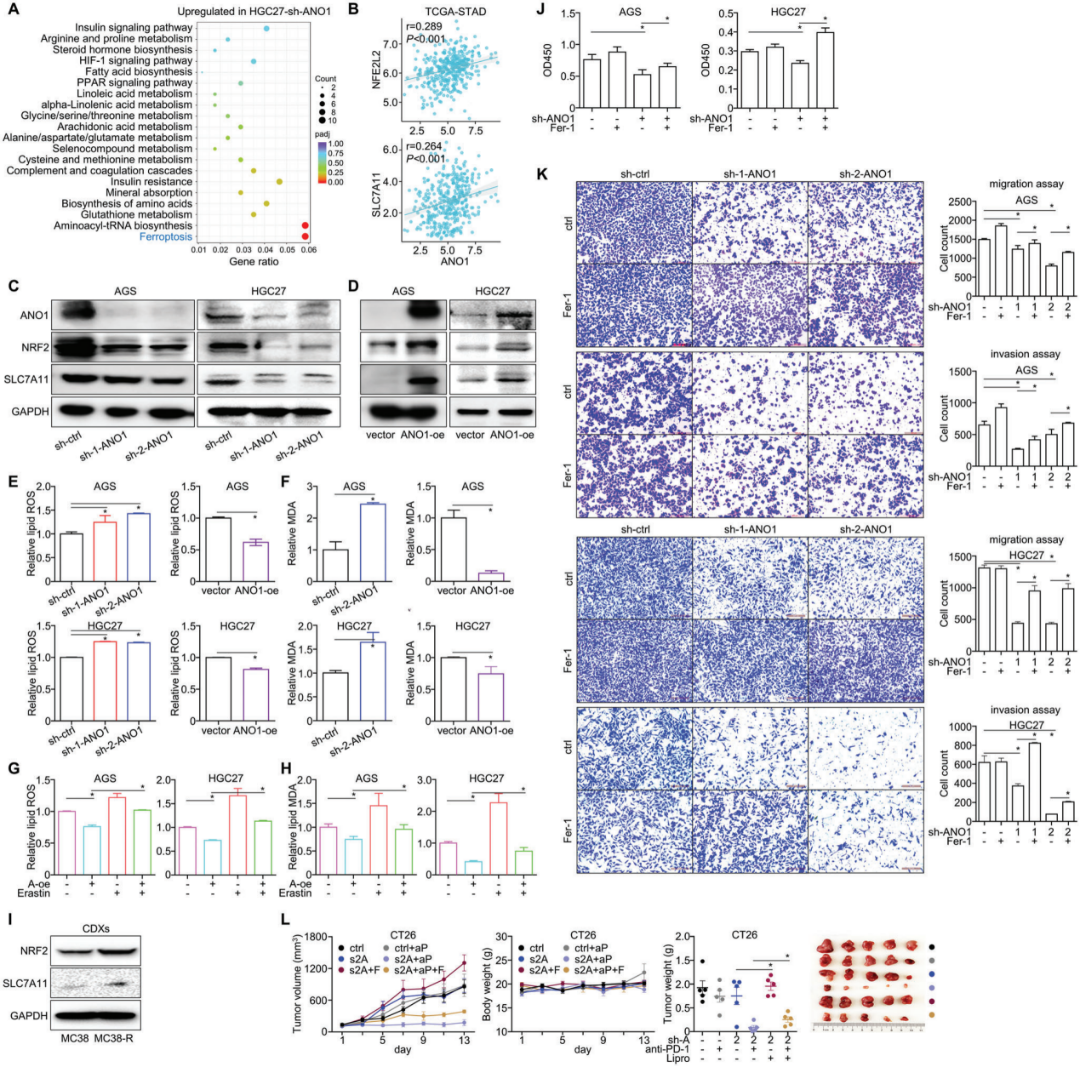
作者对ANO1敲除前、后的HGC27细胞进行了RNA测序。铁死亡通路是富集度最高的信号通路(图6A)。在TCGA-STAD数据集中,抑制铁死亡的两个关键因子NRF2/SLC7A11均与ANO1呈正相关(图6B),并在ANO1敲低或过表达GC/CRC细胞的蛋白表达水平上得到了验证(图6C,D)。此外,脂质ROS(图6E)和MDA(图6F),这两种铁死亡的主要特征物质,通过ANO1敲低显著增加而通过ANO1过表达减少,并可通过铁死亡激动剂消除蛋白逆转(图6G,H)。此外,免疫治疗获得性耐药后发现铁死亡抑制因子NRF2/SLC7A11上调(图6I)。
图6. ANO1通过抑制铁死亡促进肿瘤进展,损害免疫治疗效果
7.ANO1通过激活PI3K-Akt信号通路抑制铁死亡
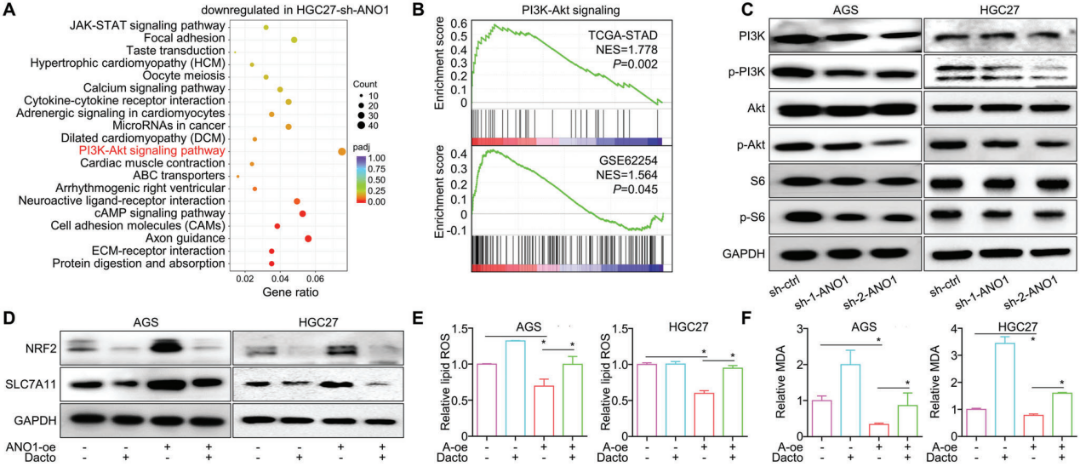
在ANO1敲除后,PI3K-Akt信号通路富集(图7A)。在TCGA的GC数据集(GSE62254)中,GSEA也表明ANO1与激活的PI3K-Akt信号通路相关(基因集M271,图7B)。在GC/CRC细胞中,表明PI3K-Akt信号通路被ANO1下调所抑制,PI3K/AKT/S6的磷酸化减少(图7C)。另一方面,过表达ANO1诱导铁死亡减速因子NRF2/SLC7A11上调(图7D),脂质ROS/MDA下调(图7E,F),并可被PI3K-Akt信号抑制剂Dactolisib撤销,说明ANO1通过激活PI3K-Akt信号抑制铁死亡。
图7. ANO1通过激活PI3K-Akt信号通路削弱铁死亡
8.ANO1介导的铁死亡诱导抑制刺激TGF-β的产生和释放,并将癌症相关的成纤维细胞招募到TIME中,产生对免疫治疗的耐药性
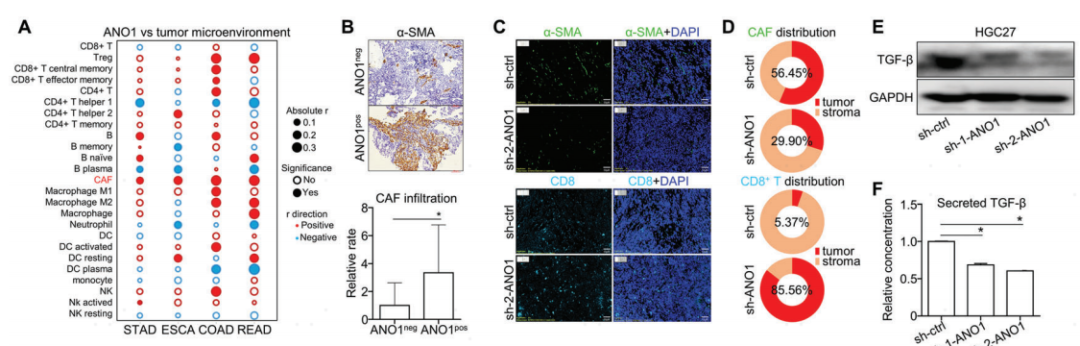
在所有关键的微环境细胞中,CAF是唯一一种在所有四种主要GI癌症的TCGA数据集上与ANO1表达一致相关的细胞类型(图8A)。根据先前分析的48例前瞻性GI癌队列中CAF标志物α-SMA的IHC染色,ANO1阳性患者的CAF相对浸润率明显高于ANO1阴性患者(图8B)。根据mIHC分析,在CT26来源的CDX组织中,ANO1敲除降低了CAF的浸润,而增强了CD8+ T细胞的浸润(图8C)。进一步分析CT26 CDX组织中细胞的空间特征,发现敲除ANO1后CAF分布的肿瘤/基质比降低,CD8+ T细胞分布的肿瘤/基质比增加(图8D)。表明ANO1敲除显著诱导CAFs从肿瘤向间质转移,CD8+ T细胞从间质向肿瘤聚集。在GC/CRC细胞中,ANO1敲低抑制TGF-β的表达和分泌(图8E,F)。
图8. ANO1通过促进癌细胞分泌TGF-β来招募CAF
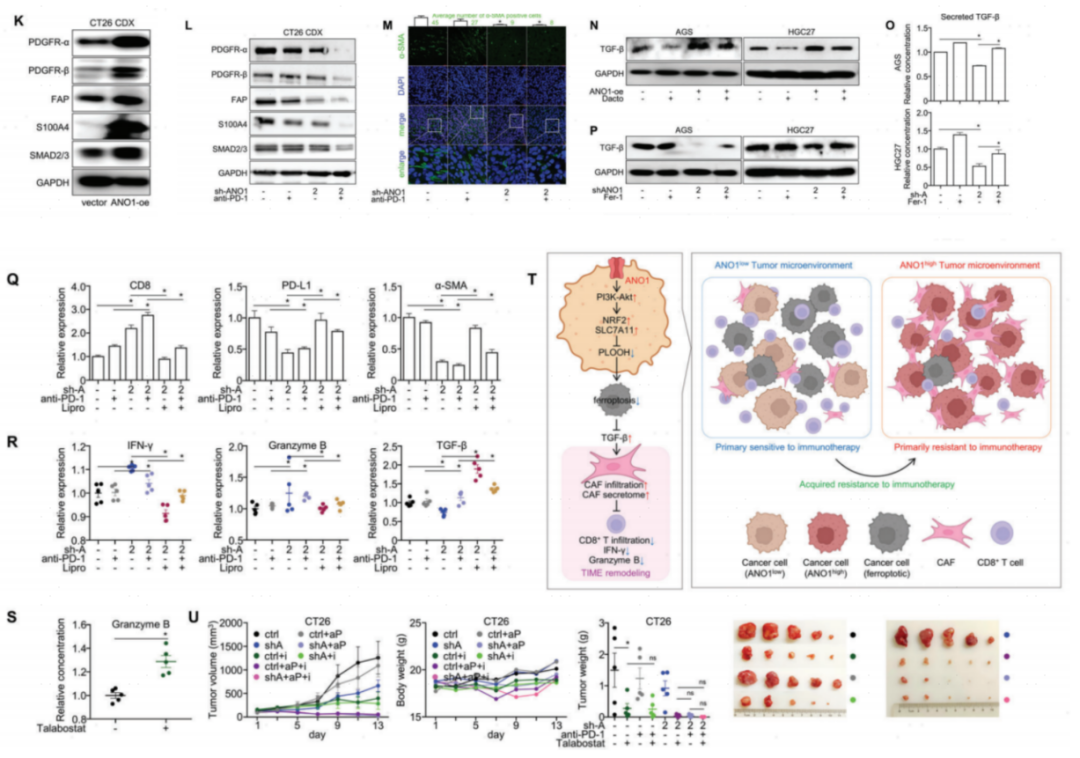
获得性免疫治疗耐药后,MC38/MC38-R CDX组织的单细胞转录组测序发现,浸润性CAFs和ANO1表达比例较高(图9G-I),CAF生物标志物(PDGFR-α/β/FAP)和TGF-β信号成员SMAD2/3蛋白表达升高(图9J)。提示抗PD-1免疫治疗获得性耐药是由ANO1招募的CAFs诱导的。
图9. 抗PD-1免疫治疗获得性耐药是由ANO1招募的CAFs诱导的
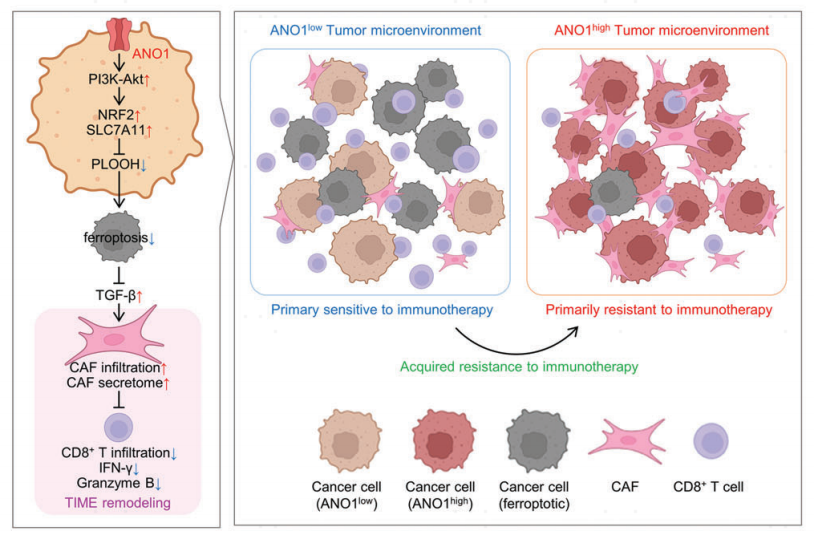
在CT26 CDX组织中,ANO1过表达后CAF生物标志物也持续升高(图10K)。结合ANO1敲除和抗PD-1抗体后,CT26 CDX组织中CAF标记物的表达(图10L)和免疫荧光染色(图10M)均显著降低。在GC/CRC细胞水平上,PI3K-Akt通路抑制剂Dactolisib抑制了ANO1过表达导致的TGF-β上调(图10N),而ANO1敲低抑制TGF-β表达/分泌的作用被铁死亡抑制剂Fer-1所恢复(图10O,P),说明TGF-β的产生受ANO1-PI3K- AKT-铁死亡信号轴调控。在CT26 CDX模型中,无论是否接受免疫治疗,ANO1敲除增加了CD8+T细胞的浸润,降低了PD-L1/α- SMA在组织中的分布。而在体内,铁死亡抑制剂利普斯他汀逆转了这些作用(图10Q)。此外,Talabostat(CAF抑制剂)增强了CT26 CDX的免疫治疗,相比于ANO1敲除/FAP抑制剂的双联疗法或ANO1敲除/抗PD-1抗体的双联疗法,ANO1敲除/抗PD-1抗体/FAP抑制剂的三联疗法可以使其抗肿瘤效果最大化(图10U)。考虑到ANO1是CAF的上游启动子,那么在ANO1抑制的情况下,靶向CAF是可以克服免疫治疗耐药性的。ANO1介导的抑制肿瘤的铁死亡会随着时间的推移增强CAF的积累,并进一步与促进ANO1诱导的免疫治疗耐药。
图10. ANO1介导的铁死亡抑制引起TGF-β的释放,并使癌相关成纤维细胞进入TIME,产生对免疫治疗的耐药性