1.相较于FBC,MBC肿瘤组织存在低水平免疫浸润
为了探究乳腺癌细胞组成,本文对6名男性乳腺癌(MBC)和13名女性乳腺癌(FBC)患者进行了scRNA-seq,通过经典marker鉴定出了上皮细胞,T细胞,B细胞,浆细胞,巨噬细胞,肥大细胞,肌成纤维细胞,肿瘤相关成纤维细胞,动脉内皮细胞,静脉内皮细胞和毛细血管内皮细胞。基于inferCNV相关分析,在上皮细胞中鉴定出了肿瘤细胞。接下来通过组间占比分析发现,MBC患者相较于FBC患者有更高的肿瘤细胞占比以及更低的T细胞占比,表明其处于低水平的免疫浸润。为了进一步验证该结果,作者基于bulk转录组数据(肿瘤纯度评分、ssGSEA特征基因打分以及解卷分析方法)以及免疫组化数据发现:MBC患者具有更高的肿瘤纯度以及更低的T/B细胞占比,这与单细胞转录组结果一致。
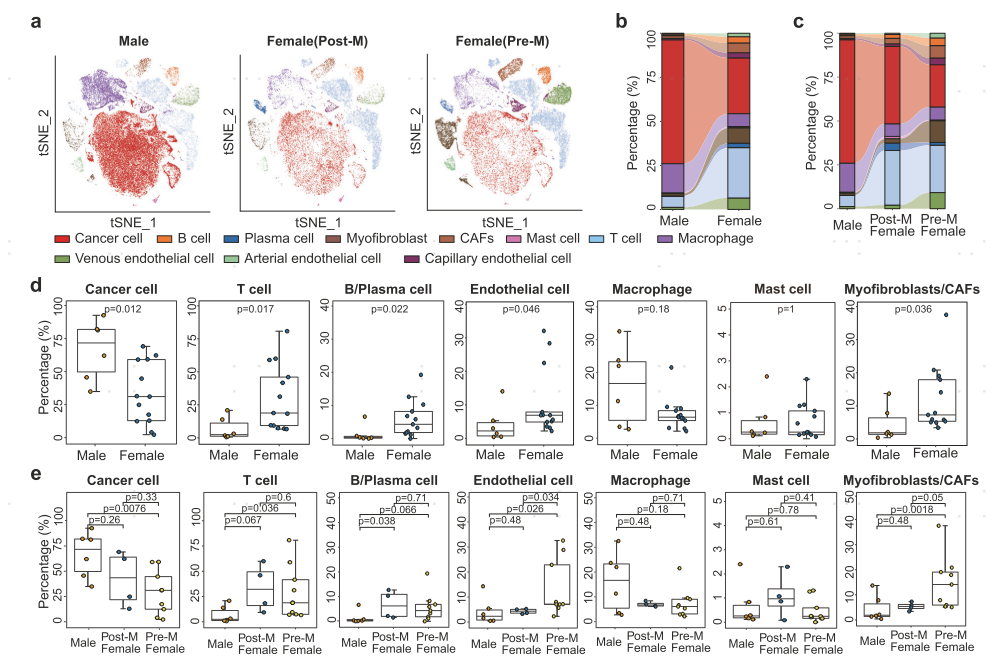
图1 相较于FBC,MBC肿瘤组织存在低水平免疫浸润
2.在MBC患者的肿瘤细胞中,代谢模块以及AR和SREBF1介导的下游调控基因显著激活
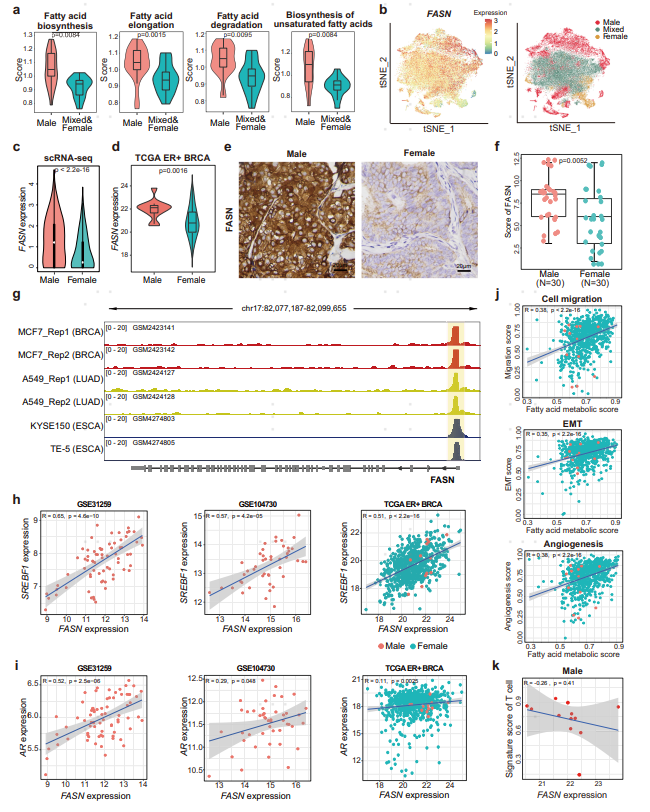
为探究MBC/FBC患者肿瘤细胞的表达模式,作者对肿瘤细胞进一步细分,并区分出MBC(分群中MBC占比超过70%),FBC(分群中FBC占比超过50%),mixed(MBC和FBC混合)三类分群。对MBC以及FBC群进行组间差异基因分析发现,MBC中高表达脂肪代谢相关基因(例如:FASN、AZGP1)。此外,通过基因特征集合分析,MBC相较于FBC,其具有较强的转移,间充质转换(EMT),血管再生以及代谢能力。
接下来作者对MBC肿瘤细胞亚群进行转录调控分析,发现相较于其他FBC亚群,MBC中雄激素受体(AR)和甾醇调节元件结合转录因子1(SREBF1)在转录因子活性和基因表达上显著上调。既往研究表明,SREBF1作为脂质代谢的重要调节因子,可促进乳腺癌的肿瘤生长和转移,并与EMT过程高度相关。此外,通过IHC免疫组化分析同样证明了AR在MBC中显著激活。
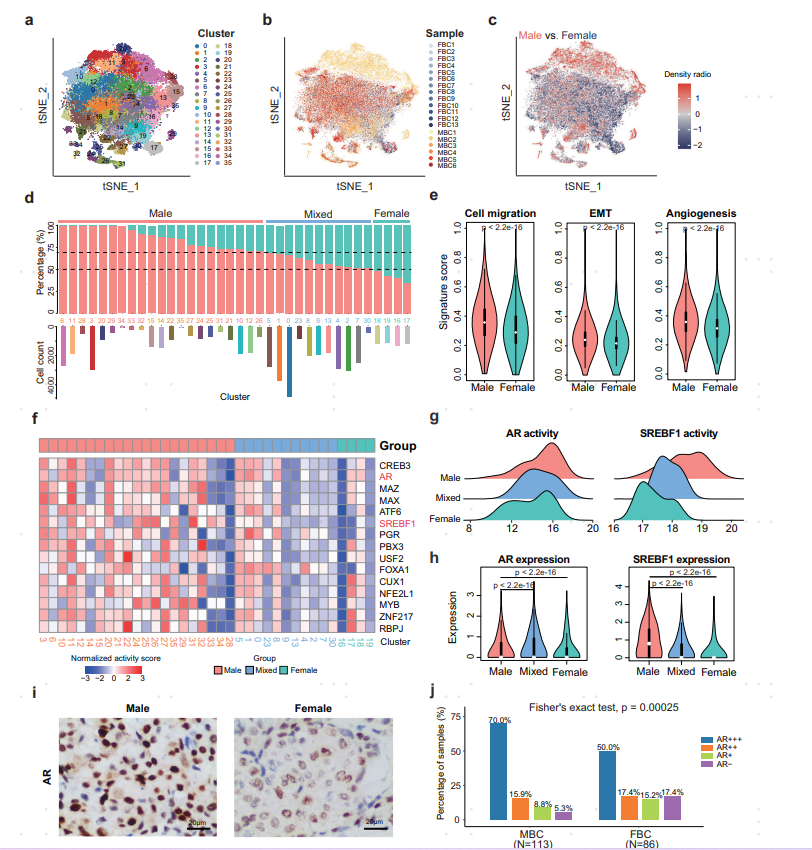
图2 在MBC患者的肿瘤细胞中,代谢模块以及AR和SREBF1介导的下游调控基因显著激活
3.激活的脂肪酸代谢与MBC的转移和低免疫浸润有关
利用代谢相关途径对肿瘤细胞进行打分,结果表明脂肪酸代谢相关途径在MBC患者肿瘤细胞激活,这其中包括脂肪酸生物合成、脂肪酸延伸/降解以及不饱和脂肪酸的生物合成。基于单细胞转录组数据,发现FASN(脂肪酸合酶)在MBC肿瘤亚群中高表达,bulk RNA数据以及IHC数据进一步验证了相关结果。
更值得注意的是,基于三个细胞系(乳腺癌/肺腺癌/食管癌)Chip-seq数据分析,发现FASN介导的脂肪酸代谢途径由转录因子SREBF1调控。同时,作者还发现脂肪酸代谢评分与细胞增殖、上皮间充质转换(EMT)、血管生成呈现正相关,表明脂肪酸代谢可能促进乳腺癌的转移;而FASN的表达与肿瘤纯度呈现正相关,与T/B细胞浸润得分呈现负相关,提示FASN的高表达与免疫排斥有关。为了进一步探究FASN基因,作者利用TCGA的泛癌数据,通过区分性别的生存预后分析发现FASN高表达组患者,尤其是在男性患者组中预后较差,提示FASN可能是多癌种中男性患者的潜在治疗靶点。
图3 激活的脂肪酸代谢与MBC的转移和低免疫浸润有关
4.MBC和FBC之间T细胞亚群的不同功能特征
本文对T细胞进行亚群细分。联合单细胞转录组以及scTCR数据,发现CD8+ CAPG+和 CD8+ IFIT1+在MBC中显著扩增,且CD8+T中p38 MAPK通路评分显著升高,表明MBC肿瘤微环境中T细胞处于功能失调状态。进一步分析发现MBC中的CD8+T显著富集到脂质代谢途径,结合前期相关研究报道,推测脂质代谢参与了T细胞功能失调。在FBC肿瘤微环境中则发现大量细胞毒性T细胞marker的表达,以及富集到免疫调节细胞因子介导的多种途径。上述结果进一步明确MBC和FBC肿瘤组中的T细胞功能存在差异。

图4 MBC和FBC的T细胞功能存在差异
5.具有高水平脂肪酸代谢的KRT8+ T细胞在MBC微环境中富集
通过分析发现,KRT8+ T细胞在MBC中特异性富集,上皮细胞标记物在MBC的T细胞中表达显著。为了验证KRT8+ T细胞在MBC患者中的存在,作者还纳入了已发表公共数据中的(Wu et al.’s)FBC样本进行验证分析,结果表明KRT8在MBC样本中的T细胞(超50%占比)高度表达。此外,为了排除低质量细胞、双细胞或者实验过程引起的细胞污染和细胞应激反应,作者采用了三种策略进行逐一排除,包括:①使用不同的阈值来限制每个单细胞内表达基因的数量,从1500到5000;②使用CellBender去除污染,Scrublet和DoubletFinder去除双细胞;③利用GSEA分析排除线粒体,核糖体,热休克蛋白基因的功能富集。后续的免疫荧光染色以及细胞流式分选实验也证明了此类细胞的存在。
为了进一步探究KRT8+ T的功能,作者进行了富集分析,发现CD3E+ KRT8+ T细胞中脂肪酸代谢显著激活,但细胞毒性能力下降,进一步证实了MBC患者脂质代谢与T细胞功能失调的相关性。
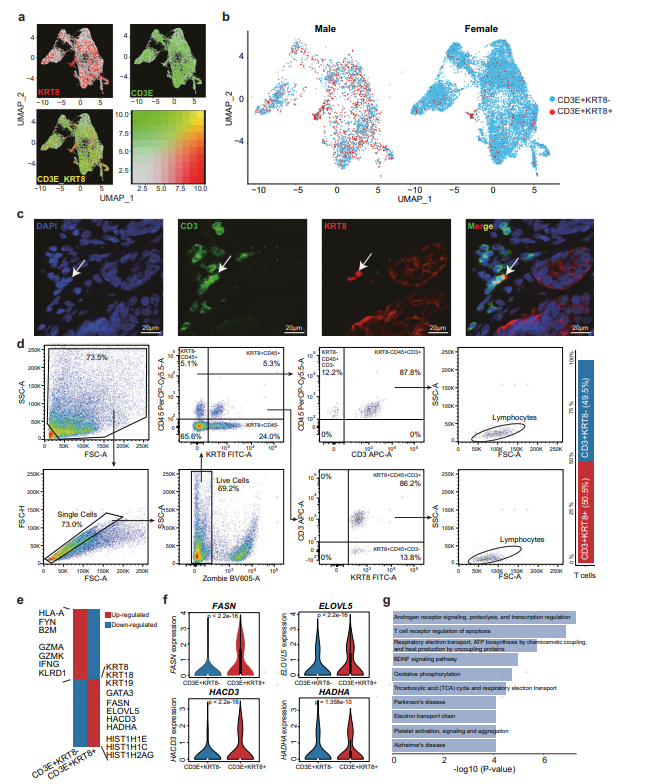 图5 具有高水平脂肪酸代谢的KRT8+ T细胞在MBC微环境中富集
图5 具有高水平脂肪酸代谢的KRT8+ T细胞在MBC微环境中富集
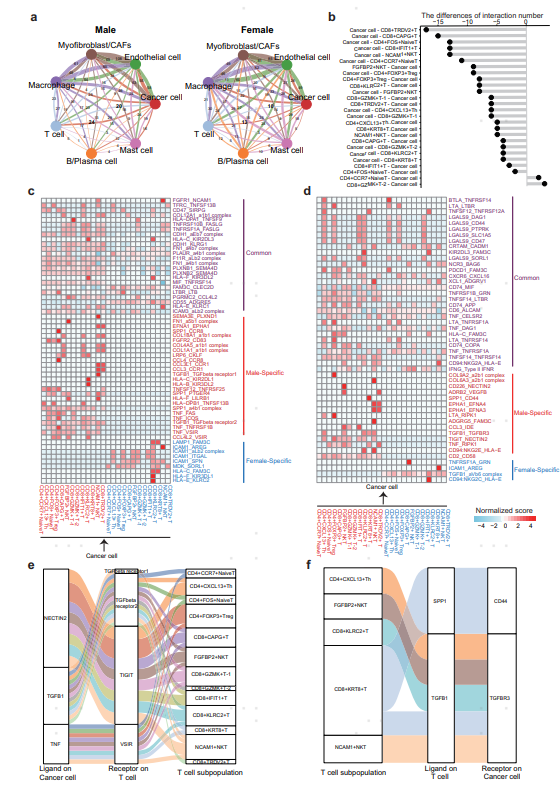
6.癌细胞与T细胞之间的通讯参与了MBC的免疫抑制
最后,作者对MBC和FBC样本中不同细胞类型之间的细胞通讯状态进行了分析,以确定免疫微环境中的细胞交互差异。结果显示,MBC样本中癌细胞和T细胞之间的相互作用数大约是FBC样本的两倍。与FBC样本相比,MBC中的大多数T细胞亚型与癌细胞有更多的相互作用。在MBC样本的癌细胞和T细胞中,TIGIT-NECTIN2、VSIR-TNF等受配体对的相互作用显著激活,提示相关互作对在免疫抑制和肿瘤进展中发挥着重要作用。
图6 癌细胞与T细胞通讯参与了MBC的免疫抑制