项目文章Cancer Discovery | 单细胞测序揭秘胰腺癌免疫治疗靶点RIPK2
发布时间:2023-11-22 10:53:11
2023年10月,浙江大学基础医学院刘云华团队、浙江大学附属第二医院王达团队、浙江大学附属妇产科医院钱俊斌团队及四川大学华西医院姜红团队在Cancer Discovery期刊上发表了题为“Receptor-interacting protein kinase 2 is an immunotherapy target in pancreatic cancer”的研究成果。该研究应用体内CRISPR筛选确定RIPK2是免疫逃避的关键驱动因素,并通过新格元GEXSCOPE®单细胞转录组测序(scRNA-Seq)技术发现CD8+T细胞是PDAC肿瘤免疫微环境(TME)中受RIPK2缺陷影响最严重的免疫细胞类型,随后发现用基因敲除或小分子抑制剂靶向RIPK2可增强PDAC对免疫检查点阻断(ICB)治疗敏感性。研究结果揭示了受体相互作用蛋白激酶2是胰脏癌的免疫治疗靶点,为PDAC的新型联合疗法(RIPK2抑制联合抗PD-1免疫疗法)提供了理论依据。
下面和元小新一起来看看吧~
研究背景
胰腺导管腺癌(PDAC)是一种高度侵袭性和致命性的恶性肿瘤,平均5年总生存率低于10%。在过去的十年中,免疫疗法,特别是免疫检查点阻断(ICB),已被证明可以增强T细胞介导的免疫反应。然而最近的临床试验报告显示,由于遗传异质性以及高度促纤维增生和免疫抑制的肿瘤微环境(TME),PDAC对单药ICB和双药ICB几乎具有一致耐药性。亟待找到新的可操作的免疫治疗靶标以提高PDAC的治疗效果。该研究利用体内CRISPR筛选,结合单细胞转录组测序技术以及各种体内体外验证实验,发现RIPK2可能是一个以前未被重视的免疫治疗靶点,将RIPK2抑制与ICB治疗相结合成为PDAC患者潜在的新型免疫治疗策略。
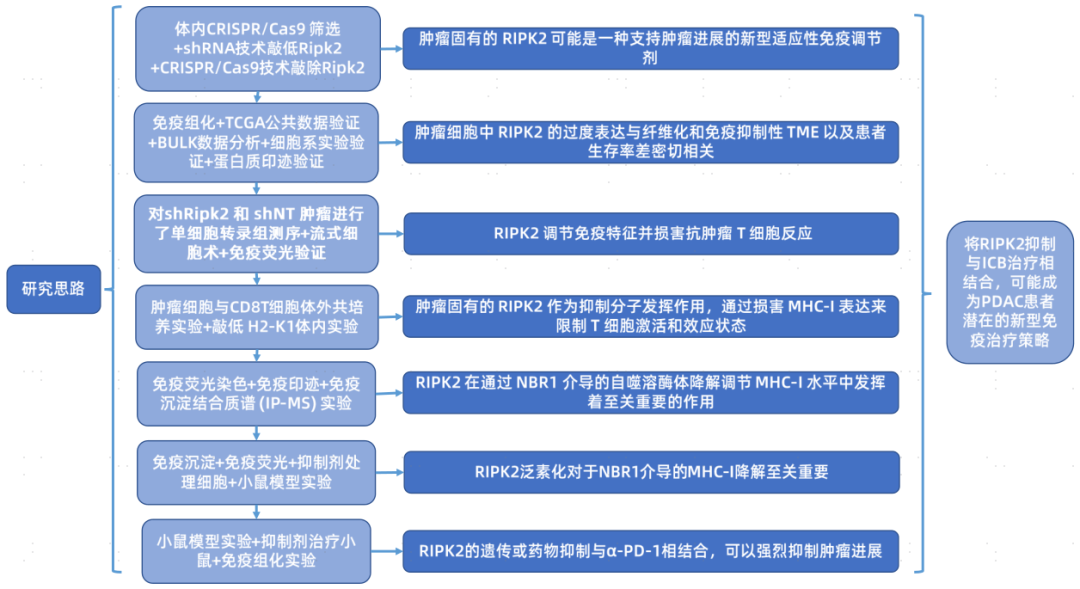
思维导图
研究结果
1.体内CRISPR筛选确定RIPK2为免疫逃避的关键驱动因素
为了系统地发现肿瘤内在决定因素(其缺失可增强抗肿瘤免疫力),作者使用原位小鼠模型进行了体内功能性CRISPR/Cas9筛选。发现在37只免疫活性小鼠的KPCmut肿瘤中(相对于裸鼠),Ripk2(编码一种激酶)成为排名最高的缺失基因。
在验证实验中,作者在三种小鼠PDAC细胞系(KC6141、KPCmut和KPCflox)中用短发夹RNA(shRNA:shRipk2-1和shRipk2-2)从基因上敲低了Ripk2。将这些细胞平行接种到免疫缺陷小鼠和同基因野生型宿主中。作者观察到,RIPK2的基因缺失导致携带shRipk2和乱序(shNT)KC6141肿瘤的裸鼠肿瘤生长适度减少,并且Ripk2敲低显著损害了免疫活性小鼠的肿瘤生长并延长了生存期。这些结果表明肿瘤固有的RIPK2可能是一种支持肿瘤进展的新型适应性免疫调节剂。
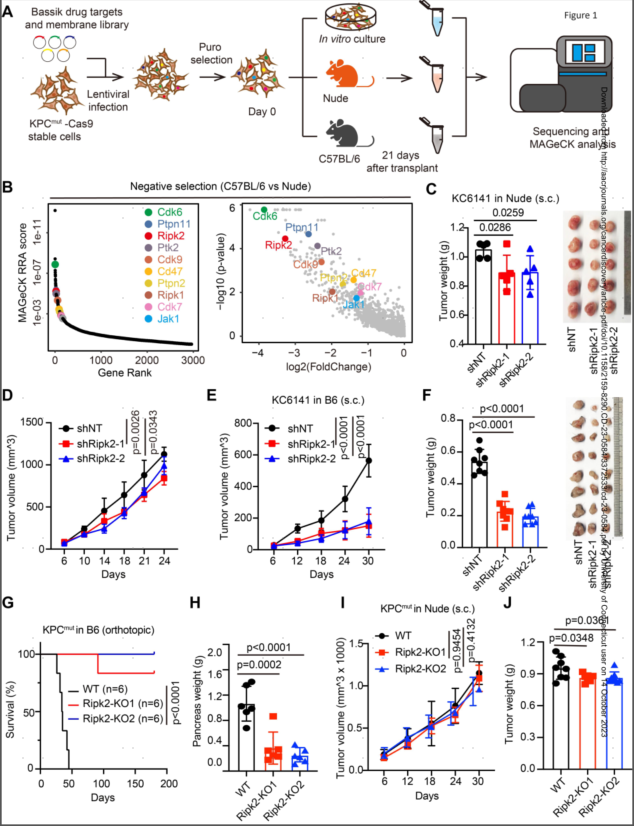
图1.体内CRISPR筛选将RIPK2确定为免疫逃避的关键驱动因素
2.RIPK2在人类PDAC中过度表达,与患者生存率低相关
鉴于RIPK2在驱动PDAC免疫逃避方面的显著作用,作者通过免疫组织化学(IHC)在包含60个PDAC肿瘤和10个正常胰腺组织的人体组织微阵列(TMA)中评估了RIPK2的表达。与正常胰腺上皮相比,RIPK2在早期胰腺上皮内瘤病变(PanIN)中可检测到,在晚期PanIN中适度上调,在PDAC病变中大幅升高。对公共数据集的生物信息分析发现,与正常胰腺组织相比,其表达显著增加,与较差的生存率相关。转录组分析表明,RIPK2表达在基底样肿瘤中富集,并与活化的基质亚型相关。为了确定RIPK2过表达是否与促纤维增生和免疫抑制性TME相关,作者评估了人PDAC组织阵列中的胶原沉积和肿瘤浸润CD8+T细胞(IHC)。结果显示,肿瘤细胞中高水平的RIPK2与较少的CD8+T细胞浸润有关,且与较高水平的胶原沉积有关。总之,这些数据表明肿瘤细胞中RIPK2的过度表达与纤维化和免疫抑制性TME以及患者生存率差密切相关。
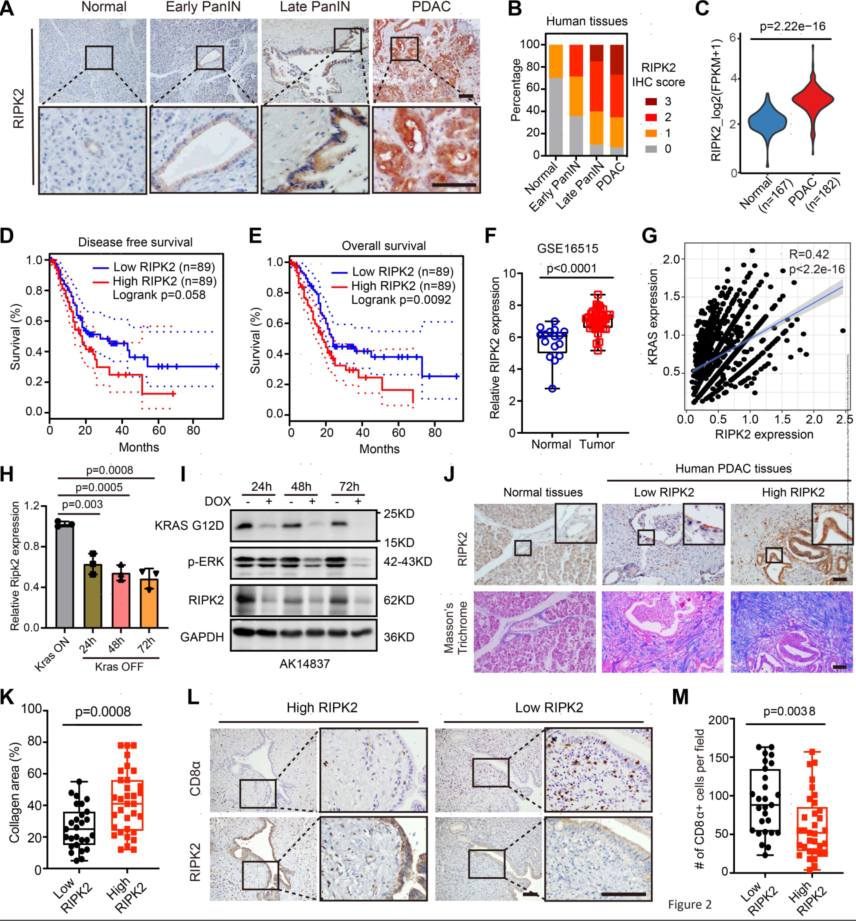
图2.RIPK2在人类PDAC中过度表达,与患者生存率低相关
3.RIPK2调节免疫特征
为揭示肿瘤固有的RIPK2缺失导致的免疫逃避机制,作者对shRipk2和shNT肿瘤进行了单细胞转录组测序。联合注释后观察到shRipk2肿瘤中淋巴细胞簇显著扩张,粒细胞簇减少。对两组的T细胞和自然杀伤(NK)细胞进行聚类分析,鉴定出14个亚簇,shRipk2肿瘤中CD8+T细胞(TEX细胞)和效应/记忆CD8+T细胞(TEM细胞)比例急剧增加。拟时序轨迹分析显示,TN与TEM和TEX细胞连接,然后分支成两个不同的轨迹,形成细胞毒性CD8+T细胞和记忆/干T细胞。从发育轨迹上可见来自shRipk2肿瘤的CD8+T细胞表达效应和细胞毒活性相关基因的水平显著高于shNT肿瘤。GSEA分析发现来自shRipk2肿瘤的CD8+T细胞富集了与T细胞活性相关途径。
作者使用流式细胞术验证了shRipk2肿瘤中CD8+T细胞密度和功能的增强。使用免疫荧光染色证实与shNT对照相比,终末期shRipk2KPCmut和KPCflox肿瘤中肿瘤浸润CD8+T细胞显著增加。来自shRipk2肿瘤的很大一部分CD4+和CD8+T细胞表现出效应表型,只有少数是幼稚的表型。更重要的是,基于Ki67水平,肿瘤细胞中的RIPK2消除不仅促进了肿瘤内CD8+T细胞增殖,而且增加了存在于肿瘤浸润性CD8+T细胞中细胞溶解颗粒和细胞因子的产生,表明这些细胞的功能性和活性状态有所增加。
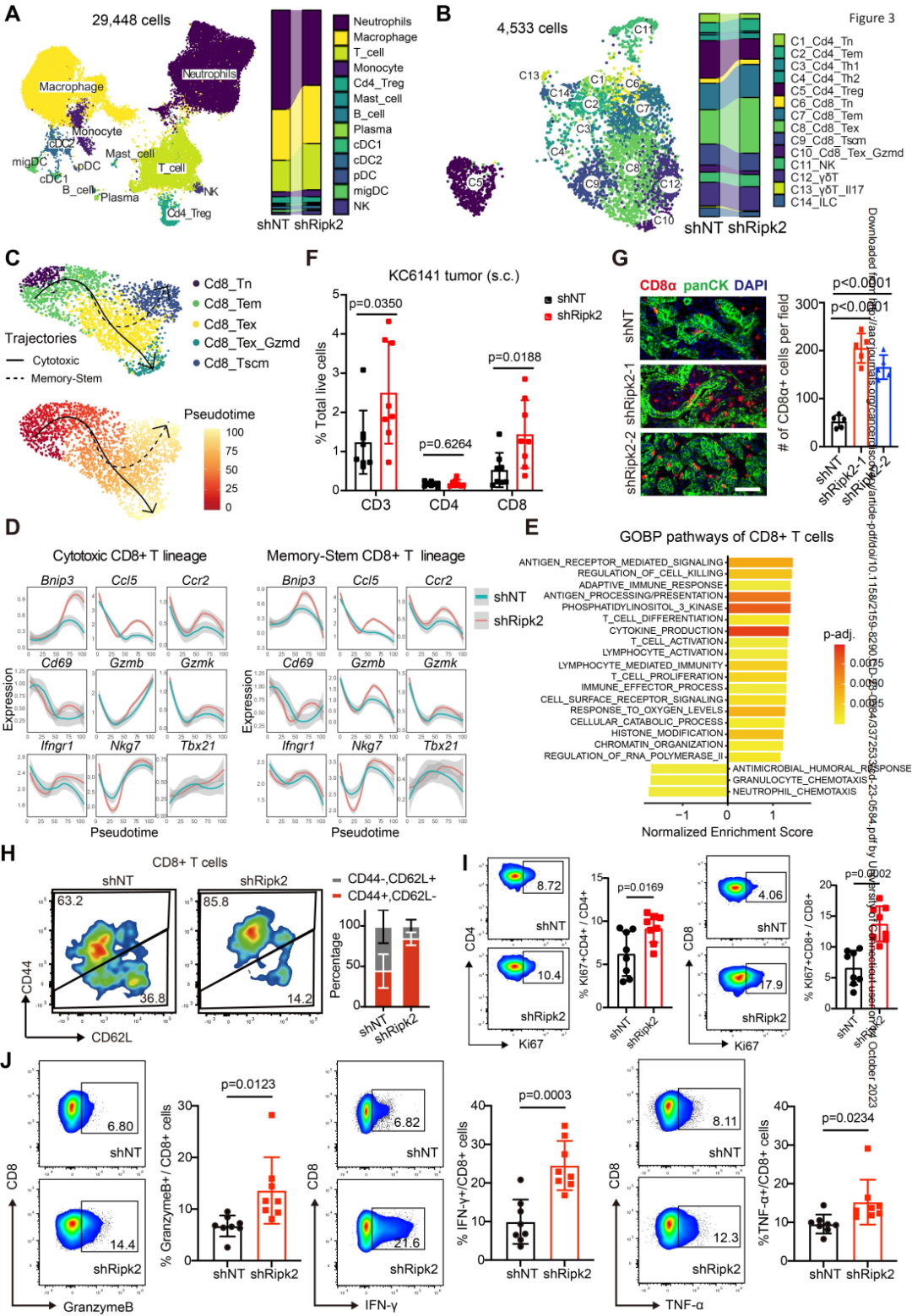
图3.RIPK2调节免疫特征并损害抗肿瘤T细胞反应
4.RIPK2通过损害抗原呈递来限制CD8+T细胞状态
为了确定肿瘤细胞中RIPK2的表达是否直接影响CD8+T细胞的激活和效应状态,作者将表达卵清蛋白(OVA)的KPCmut细胞与CD8+T共培养,发现OT-IT细胞表现出CD69以及TNF-α、IFN-γ和颗粒酶B的强表达。此外,当与活化的OT-IT细胞共培养时,表达shRipk2的细胞比对照KC6141-OVA或KPCmut-OVA细胞更容易受到CD8+T细胞介导的杀伤。值得注意的是,这些效应是MHC-I特异性的,因为H-2Kb-SIINFEKL阻断抗体抑制OT-I细胞增殖,表明MHC-I介导的抗原呈递可能参与该功能。
鉴于CD8+T细胞激活是通过TCR与抗原衍生肽-MHC-I复合物结合介导的,作者假设RIPK2可能影响肿瘤细胞表面的抗原呈递。与乱序对照相比,表达shRipk2的小鼠PDAC细胞中与SIINFEKL结合的H-2Kb分子的染色显著增加。敲除RIPK2也提高了多种人PDAC细胞系中I类HLA的表面水平。与shNT对照相比,原位植入的表达shRipk2的KPCmut和KPCflox肿瘤细胞上的H-2Kb而非PD-L1的表面水平也有所增加。H2-K1敲低导致体内MHC-I表达丧失,挽救了shRipk2肿瘤生长迟缓,并减少了shRipk2表达肿瘤中CD8+T细胞的数量。总的来说,这些结果表明肿瘤固有的RIPK2作为抑制分子发挥作用,通过损害MHC-I表达来限制T细胞激活和效应状态。
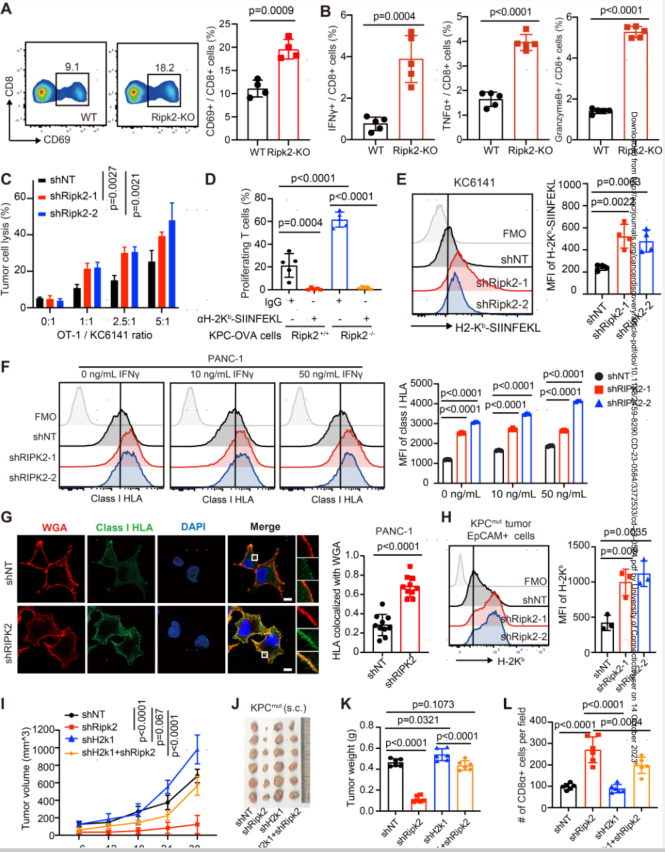
图4.RIPK2通过损害抗原呈递来限制CD8+T细胞的激活和效应状态
5.RIPK2通过NBR1促进MHC-I转运至溶酶体
接下来作者探讨了RIPK2耗尽后PDAC肿瘤细胞中MHC-I表达如何增强。在shNT对照和shRIPK2细胞中检测到HLA-A/B/C以及MHC-I抗原加工和呈递(APP)转录本的水平相当,表明RIPK2可能调节MHC-I蛋白稳定性而不是mRNA转录本。实验测试表明肿瘤细胞中RIPK2的消除会损害MHC-I降解,导致MHC-I蛋白稳定性升高。
因为膜蛋白,包括MHC-I,通常在溶酶体和/或蛋白酶体中运输和降解,所以作者用V-ATP酶抑制剂巴弗洛霉素(BafA1)处理PANC-1和CFPAC-1细胞以抑制溶酶体功能,结果表明RIPK2通过溶酶体介导的降解在调节I类HLA水平方面发挥着至关重要的作用。此外,作者对PANC-1和HPAC-1细胞中的LAMP1(溶酶体标记物)和LC3(自噬体标记物)进行了免疫荧光染色,发现RIPK2的缺失显著降低了I类HLA在溶酶体和自噬体中的定位。这表明RIPK2的消除可能主要改变自噬体的合成,从而抑制MHC-I的降解。
先前的研究发现RIPK2是一种接头激酶,通过与多种蛋白质相互作用来调节先天免疫信号。为了确定其潜在相互作用,进行了免疫沉淀结合质谱(IP-MS)实验,检测到NBR1是RIPK2免疫沉淀物中最丰富的亚基之一。NBR1是一种自噬受体,已被证明可以与泛素化底物(包括MHC-I)相互作用并靶向其降解。作者推断RIPK2可能调节NBR1介导的MHC-I自噬溶酶体降解。为检验这一假设,通过免疫沉淀和免疫荧光等实验证明了RIPK2在通过NBR1介导的自噬溶酶体降解调节MHC-I水平中发挥着至关重要的作用。
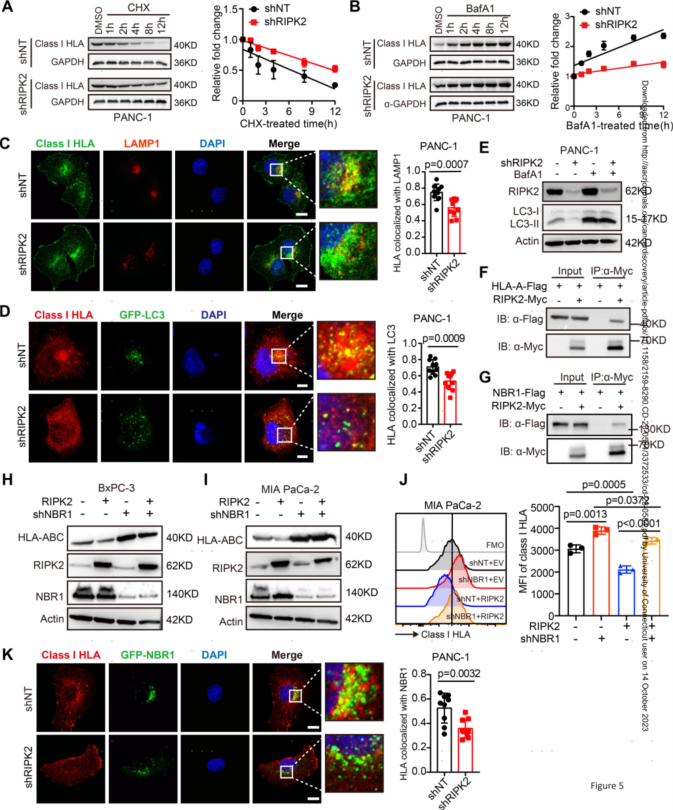
图5.RIPK2促进MHC-I通过NBR1运输至溶酶体
6.RIPK2泛素化促进NBR1介导的MHC-I降解
RIPK2具有N端激酶结构域(KD)和C端caspase激活和募集结构域(CARD)。为了研究RIPK2如何调节NBR1介导的MHC-I降解,生成了一系列缺乏CARD结构域或已知磷酸化(D146)和泛素化(K410/538)位点的突变体。免疫沉淀分析表明,RIPK2与HLA-A和NBR1的功能相互作用需要RIPK2的泛素化。具体来说,K410/538R突变体几乎完全失去了与HLA-A和NBR1的相互作用,而ΔCARD和D146N突变体部分保留了它们的结合活性。在PDAC细胞中检测到泛素化RIPK2水平明显高于胰腺导管上皮细胞。此外,与野生型RIPK2对照相比,表达shRIPK2的PANC-1细胞中K410/538R突变体的异位表达导致RIPK2泛素化显著降低。在表达shRIPK2的人PDAC细胞中,K410/538R突变体的异位表达对MHC-I在细胞表面的呈递没有显著影响,而部分结合缺陷形式的RIPK2则减少了MHC-I的呈递。此外,用GSK583(一种临床前RIPK2选择性抑制剂,RIPK2i)或ponatinib(一种FDA批准的多靶点抑制剂)治疗可有效消除人和小鼠PDAC细胞中的RIPK2泛素化。使用GSK583或ponatinib对RIPK2进行药理抑制显著增加了MHC-I在多个PDAC细胞表面的表达。这些结果表明K410/538中的RIPK2泛素化对于NBR1介导的MHC-I降解至关重要。
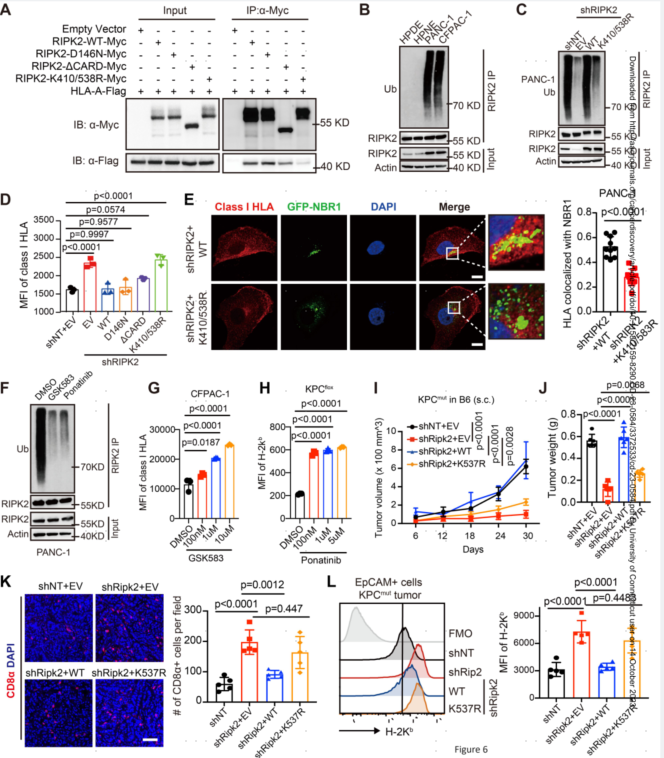
图6.RIPK2泛素化促进NBR1介导的MHC-I降解
7.RIPK2消融增强PD-1阻断的功效
尽管RIPK2消融后抗肿瘤免疫反应重新活跃,但CD8+T细胞中PD1检查点的表达水平仍较高。因此,作者将携带原位shNT或shRipk2KPCmut肿瘤的小鼠用α-PD-1或IgG对照进行治疗。与对照组相比,单独消除Ripk2可显著减少肿瘤生长并延长预期寿命,单独使用α-PD-1疗法对肿瘤进展没有影响,而将Ripk2消融与α-PD-1相结合可显著提高总体存活率。同样,携带KPCmut肿瘤的小鼠接受α-PD-1加GSK583(10mg/kg/天)(一种临床前RIPK2选择性抑制剂)治疗,结果显示总生存期显著延长,肿瘤负荷减少。为了进一步确定研究结果的临床相关性,作者通过连续一系列的人类PDAC组织切片评估了RIPK2和MHC-I蛋白的表达。IHC分析显示,异常的RIPK2表达与MHC-I的表达呈负相关。这些结果表明,RIPK2的遗传或药物抑制与α-PD-1相结合,可以强烈抑制肿瘤进展,为免疫学“冷”PDAC肿瘤的潜在治疗策略铺平道路。
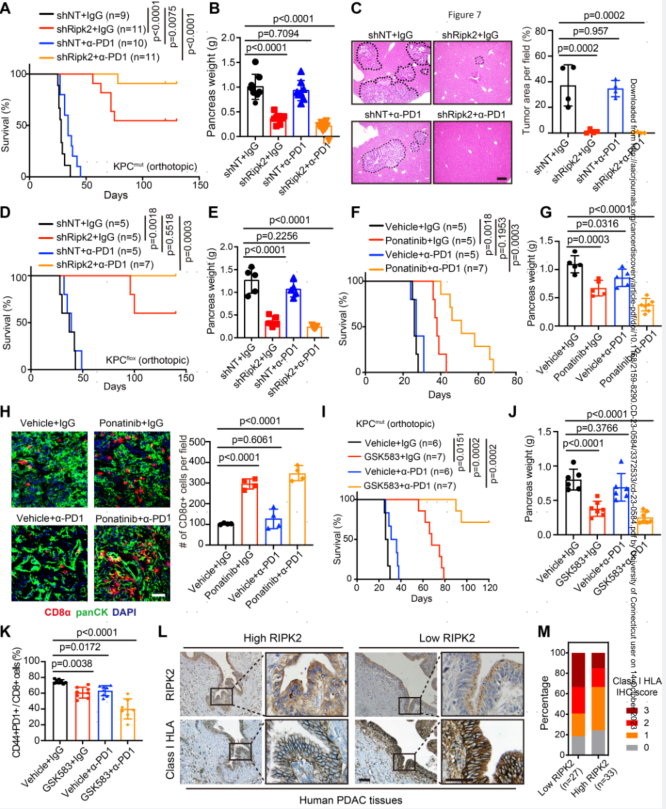
图7.RIPK2消融增强了PD-1阻断的功效
结论
该研究进行了体内CRISPR筛选,并确定RIPK2是免疫逃避的关键驱动因素,也是PDAC免疫疗法增敏有希望的候选者。用基因敲除或小分子抑制剂靶向RIPK2可使PDAC对ICB治疗敏感,从而导致完全或部分消退并延长生存期。功能和机制研究表明,肿瘤内在的RIPK2消融破坏了促纤维增生性TME,并通过消除NBR1介导的自噬溶酶体降解来恢复MHC-I的表面水平。因此,将RIPK2抑制与ICB治疗(例如抗PD-1治疗)相结合,可能成为PDAC患者潜在的新型免疫治疗策略。
参考文献
Sang W, Zhou Y, Chen H, et al. Receptor-interacting protein kinase 2 is an immunotherapy target in pancreatic cancer. Cancer Discov. 2023 Oct 12. doi: 10.1158/2159-8290.CD-23-0584.







