项目文章 | 单细胞多样同测揭示Kindlin-2在乳腺发育与泌乳功能中的作用机制
发布时间:2023-11-30 14:32:15
北京大学基础医学院、北京大学国际癌症研究院张宏权/战军研究团队在Cell Death & Disease杂志上在线发表题为“Kindlin-2 in myoepithelium controls luminal progenitor commitment to alveoli in mouse mammary gland”的文章。该研究采用5种基因修饰小鼠揭示了乳腺发育异常和泌乳功能障碍的新分子机制。
新格元在该研究中承担了CLindex®单细胞转录组多样同测实验相关的工作。
下面和元小新一起来看看吧~
研究背景
母乳喂养是婴儿的规范喂养过程,不但可以提高婴儿的生存率还能增进宝妈妈的健康。然而,全世界有5-15%的妇女患有乳腺发育异常和泌乳功能障碍。严格的信号通路调控是保证胚胎期和出生后正常的乳腺发育及泌乳功能的重要基础。其中乳腺干细胞中Notch1的激活对于促进管腔细胞规范化和驱动向单能雌激素受体阴性管腔祖细胞的渐进过渡至关重要。在妊娠和哺乳的最后三个月,Notch通路的激活受到抑制。但是Notch通路配体被调节的机制还不清楚。Kindlin-2(由FERMT2基因编码)这一整合素相互作用蛋白被发现在中胚层及其来源组织器官中高表达,尽管大量的研究报道Kindlin-2在肿瘤中通过蛋白质相互作用调控Wnt、Hippo、EGFR以及TGFβ等重要信号通路,但其在乳腺发育过程中的生物学功能及作用的分子机制仍是未知。
思维导图

研究结果
1.乳腺上皮特异性敲除Kindlin-2抑制泌乳
作者首先基于前人不同时期乳腺单细胞分析结果,确定了Kindlin-2主要在基底和肌上皮细胞中表达(图1A)。为了充分探索Kindlin-2在乳腺中的功能,作者使用Cre-LoxP系统构建了肌上皮细胞特异性(K14-Cre+; Kindlin-2flox/flox)和腔上皮细胞特异性(MMTV-Cre+; Kindlin-2flox/flox)Kindlin-2基因敲除小鼠。采用多色免疫荧光染色法探讨了Kindlin-2在小鼠肌上皮细胞中的定位和表达(图1B)。与先前报道的单细胞情况一致,Kindlin-2在肌上皮细胞(CK5标记)中表达水平更高,并且在(K14-Cre+; Kindlin-2flox/flox)组中明显被敲除(图1C)。
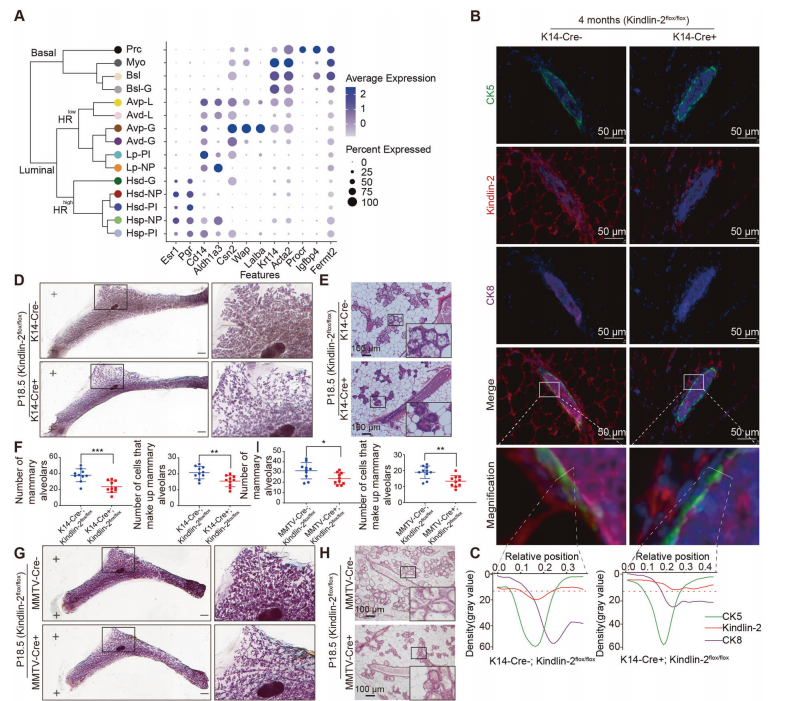
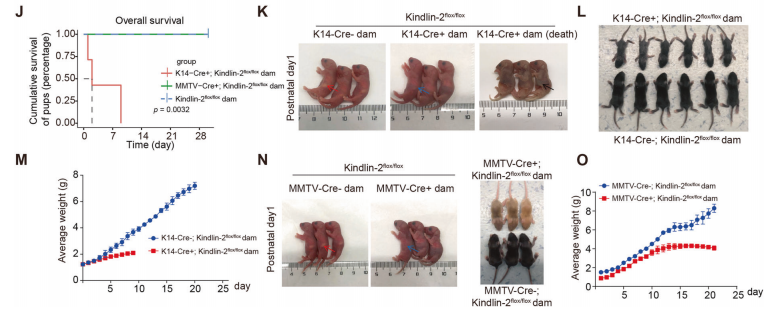
图1.乳腺上皮特异性敲除Kindlin-2抑制雌性小鼠泌乳
对小鼠妊娠期乳腺的分析表明Kindlin-2在腺泡形态发生中起重要作用。在(K14-Cre+; Kindlin-2flox/flox)中,Kindlin-2缺失的小鼠在妊娠18.5天时腺泡的形成明显受损,表现为存在小而稀疏分布的腺泡(图1D,E),并且(K14-Cre+; Kindlin-2flox/flox)组的腺泡数量和构成腺泡的细胞数量明显减少(图1F)。在(MMTV-Cre+; Kindlin-2flox/flox)组中,Kindlin-2缺乏的小鼠也在妊娠18.5天时腺泡的形成明显受损(图G-I)。这些结果表明,Kindlin-2的缺失会影响妊娠期小鼠乳腺的腺泡形成。
哺育幼崽的能力是评价乳腺功能最重要的指标之一。作者发现所有由(K14-Cre+; Kindlin-2flox/flox)产生的幼崽在9天内死亡,而(Kindlin-2flox/flox)和(MMTV-Cre+; Kindlin-2flox/flox)产生的幼崽存活(图1J)。这些结果表明,肌上皮细胞中Kindlin-2的缺失会严重损害泌乳。
基于Kindlin-2在上皮特异性敲除小鼠模型中的分布及哺乳能力的差异,作者提出Kindlin-2在肌上皮中对控制乳腺发育和泌乳功能起重要作用。
2.单细胞转录组测序(scRNA-seq)揭示了Kindlin-2缺失对妊娠晚期乳腺微环境的影响
为了揭示肌上皮细胞Kindlin-2基因敲除诱导妊娠和哺乳期疾病的机制,作者从妊娠18.5天的(K14-Cre+; Kindlin-2flox/flox)、(MMTV-Cre+; Kindlin-2flox/flox)、(Kindlin-2flox/flox)小鼠中分离了6个乳腺进行scRNA-seq分析(图2A)。质控过滤后,保留了15,719个高质量的细胞并进行了分析。根据标记基因,将细胞分为4种免疫细胞(T/NK细胞、树突状细胞、巨噬细胞和单核细胞)、1种间质细胞(成纤维细胞)和6种上皮细胞(腺泡分化细胞、腺泡祖细胞、管腔祖细胞、激素感应管腔细胞、肌上皮细胞和内皮细胞)。作者发现(K14-Cre+; Kindlin-2flox/flox)乳腺中成熟腺泡细胞较少,管腔祖细胞较少。而在(MMTV-Cre+; Kindlin-2flox/flox)中得到改善(图2C)。
为了详细探讨Kindlin-2在乳腺上皮细胞成熟和分化中的功能,对乳腺上皮细胞单独聚类分群(图2E)。利用Monocle 2构建伪时间分化轨迹图,揭示了管腔祖细胞到腺泡分化细胞的成熟过程(图2F)。作者还观察到乳汁相关基因(Cd36、Csn2、Fabp3、Fasn、Lpl和Olah)的表达水平随着成熟和终末分化逐渐增加(图2H)。
这些结果表明,肌上皮细胞中Kindlin-2的缺失会阻碍管腔祖细胞的终末分化和成熟(图2L)。
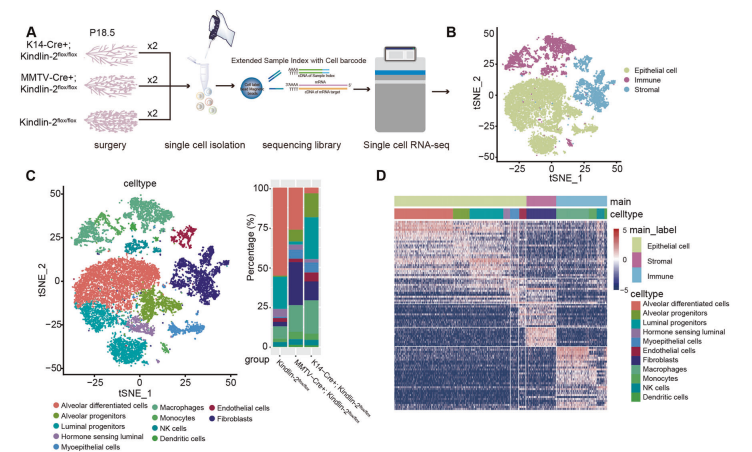
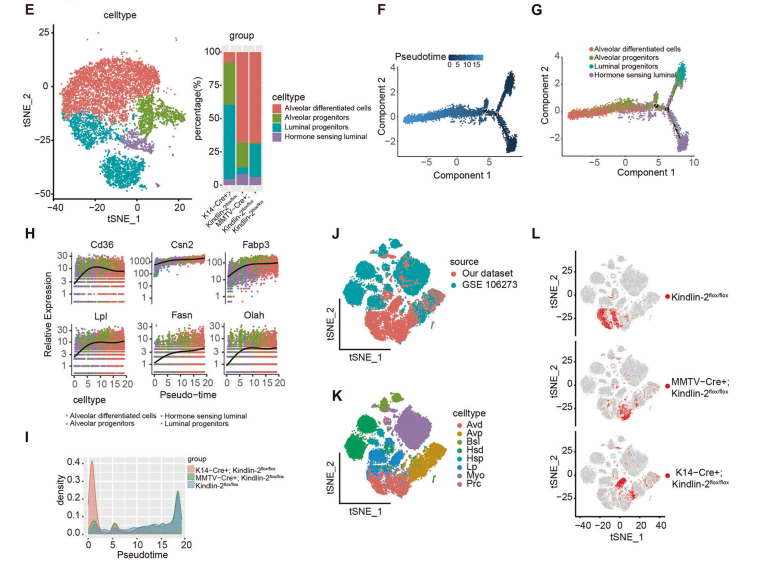
图2.scRNA-seq揭示了Kindlin-2缺失对妊娠晚期乳腺微环境的影响
3.肌上皮Kindlin-2的特异性敲除激活了管腔上皮的Notch通路
细胞间通讯对维持乳腺微环境的平衡至关重要,细胞间通讯失衡可能导致功能障碍。CellChat分析表明Notch信号的主要发送者是肌上皮细胞,而受体包括所有类型的未成熟管腔细胞(图3A)。Notch通路是介导乳腺腔细胞增殖和干性的典型途径,敲除肌上皮细胞中的Kindlin-2显著增加Notch通路的活性,这是由于经典Notch通路配体Dll1表达的升高(图3B)。Dll1主要表达于基底细胞和肌上皮细胞,而Notch1主要表达于未成熟祖细胞,其典型泌乳相关标志物水平较低,与之前的研究结果相呼应(图3C)。
为了验证Kindlin-2缺失在肌上皮细胞中引起的Notch通路的改变,作者采用多色免疫荧光染色来比较和定位小鼠妊娠18.5d时,以CK5标记的肌上皮细胞和以CK8标记的管腔细胞的Notch1和Dll1的表达(图3D)。与(Kindlin-2flox/flox)组相比,(K14-Cre+; Kindlin-2flox/flox)组肌上皮细胞中Dll1蛋白水平显著上调,Notch1在管腔细胞中的表达更高(图3E-G)。
与scRNA-seq结果一致,作者证实乳腺的(K14-Cre+; Kindlin-2flox/flox)在妊娠晚期表现出较强的Notch通路通讯和激活,该配体Dll1在肌上皮细胞高表达,而管腔细胞处于Notch1高表达的低分化状态。
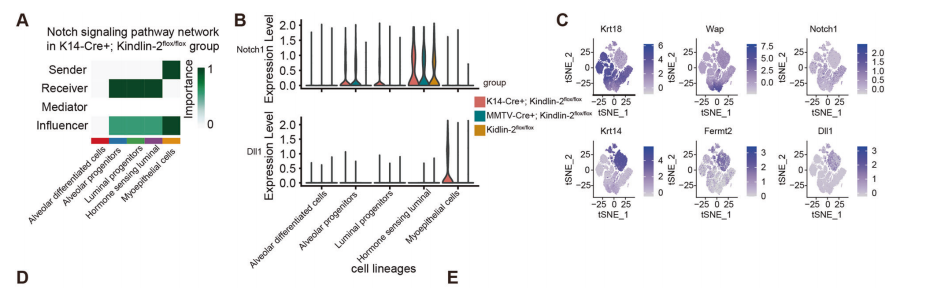

图3.肌上皮Kindlin-2的特异性敲除激活了管腔上皮的Notch通路
4.肌上皮特异性Kindlin-2敲入抑制管腔上皮Notch通路激活
为了阐明Kindlin-2在肌上皮细胞中的过度表达是否促进乳腺发育,作者构建了肌上皮特异性敲入Kindlin-2小鼠(K14-Cre+; Kindlin-2LSL/LSL)。分析发现,与(Kindlin-2LSL/LSL)组相比,(K14-Cre+; Kindlin-2LSL/LSL)组肌上皮细胞中的Dll1、管腔上皮细胞中的Notch1和Hes1的表达水平明显降低(图4A,B)。同时,(K14-Cre+; Kindlin-2LSL/LSL)组中乳腺导管厚度更大(图4C)。而(K14-Cre+; Kindlin-2LSL/LSL)组中乳腺泡的数量和构成乳腺泡的细胞数量和(Kindlin-2LSL/LSL)组是相似的。表明肌上皮中Kindlin-2的过度表达并没有显著促进腺泡的形成。
这些结果表明,过表达Kindlin-2可抑制肌上皮细胞和管腔细胞之间的Dll1/Notch1串扰,从而促进乳腺导管的发育。而在成熟阶段,Kindlin-2的内在机制是保护妊娠期乳腺的发育。腺泡的形成在(K14-Cre+; Kindlin-2LSL/LSL)和(Kindlin-2LSL/LSL)组间无明显差异(图4D-F)。

图4.肌上皮特异性Kindlin-2敲入抑制管腔上皮Notch通路激活
5.Kindlin-2与Stat3相互作用并抑制Stat3的激活
为了确定Kindlin-2在乳腺中功能的关键分子机制,作者使用DoRothEA软件评估转录因子活性(图5B),与(MMTV-Cre+; Kindlin-2flox/flox)和(Kindlin-2flox/flox)组相比,共有96个转录因子在(K14-Cre+; Kindlin-2flox/flox)组肌上皮细胞中具有较高的活性。结合先前的研究结果分析发现,Stat3和Rela可能具有结合Kindlin-2的能力(图5C)。
作者推测Kindlin-2可能通过破坏Jak和Stat3的相互作用和催化反应来调节Stat3的磷酸化和活化。因此作者检测了Stat3典型靶基因Irf1、Junb、Socs3、Fos和Hspb1的表达水平,发现它们在(K14-Cre+; Kindlin-2flox/flox)乳腺肌上皮细胞中的表达水平显著升高,这与之前对转录因子活性的分析一致(图5D)。作者证实内源性Kindlin-2可以在MDA-MB-231和HEK293T细胞中与Stat3相互作用(图5E,F)。然而,Kindlin-2的敲除只在MDA-MB-231细胞促进了Stat3在Y705和S727位点的磷酸化,说明这两个位点对MDA-MB-231细胞中Stat3的激活都很重要(图5G)。作者假设Kindlin-2和Stat3之间的调节相互作用可能主要发生在肌上皮细胞和基底细胞中,即(MMTV-Cre+; Kindlin-2flox/flox)小鼠中Kindlin-2的敲除不中断Stat3的激活。Kindlin-2的缺失增加了MDA-MB-231细胞中Stat3与抗Jak2抗体共免疫沉淀的数量(图5H)。
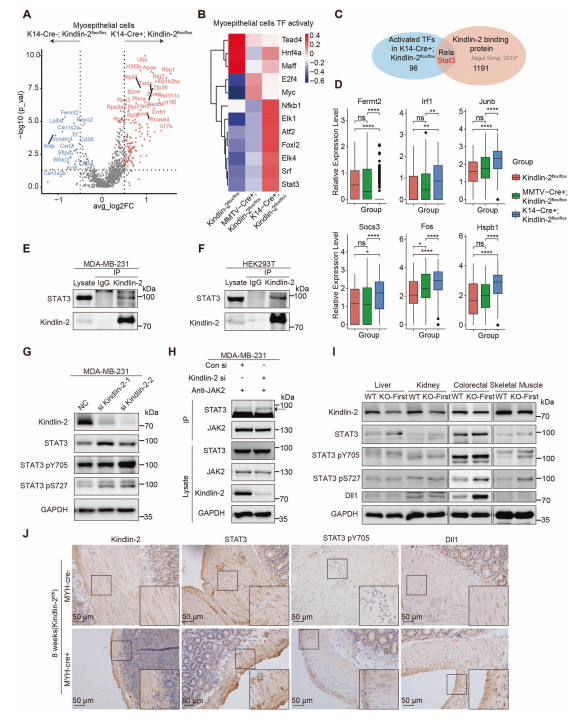
图5.Kindlin-2与Stat3相互作用并抑制Stat3的激活
为了确定Kindlin-2的缺失是否通过平滑肌组织中的共同机制促进Stat3磷酸化和Dll1的表达,作者从Kindlin-2敲除(KO-First)和WT小鼠中获得肝脏、肾脏、结直肠和骨骼肌组织。在肌肉层含有丰富平滑肌的结直肠组织中,KO-First组Dll1表达水平和Stat3磷酸化水平显著升高(图5I)。此外,作者在(Kindlin-2fl/fl; MYHcre+)小鼠(Kindlin-2在注射他莫昔芬后在平滑肌中被特异性敲除)结直肠组织中检测到Stat3磷酸化位点和Dll1的表达均较高(图5J)。
6.Kindlin-2的缺失激活Stat3并上调肌上皮中的Dll1
为了确认Kindlin-2和Stat3在乳腺微环境中的原位调节相互作用,作者使用多色免疫荧光染色鉴定了(K14-Cre+; Kindlin-2flox/flox)和(Kindlin-2flox/flox)组肌上皮细胞中Stat3的磷酸化水平。研究发现(K14-Cre+; Kindlin-2flox/flox)组肌上皮细胞表现出相对较高的Stat3表达水平,Y705和S727位点的磷酸化水平显著升高(图6A-D)。Y705和S727磷酸化水平与Dll1表达一致,特别是在(K14-Cre+; Kindlin-2flox/flox)肌上皮细胞中(图6E)。这些结果表明,肌上皮细胞Kindlin-2缺失可诱导Stat3磷酸化,磷酸化后的Stat3作为激活转录因子诱导Dll1的表达,从而原位调控Notch通路的激活。
从这些结果中,作者确定Kindlin-2是肌上皮细胞中一个重要的调节因子,通过Jak2-Stat3-Dll1-Notch1轴控制管腔细胞的成熟。
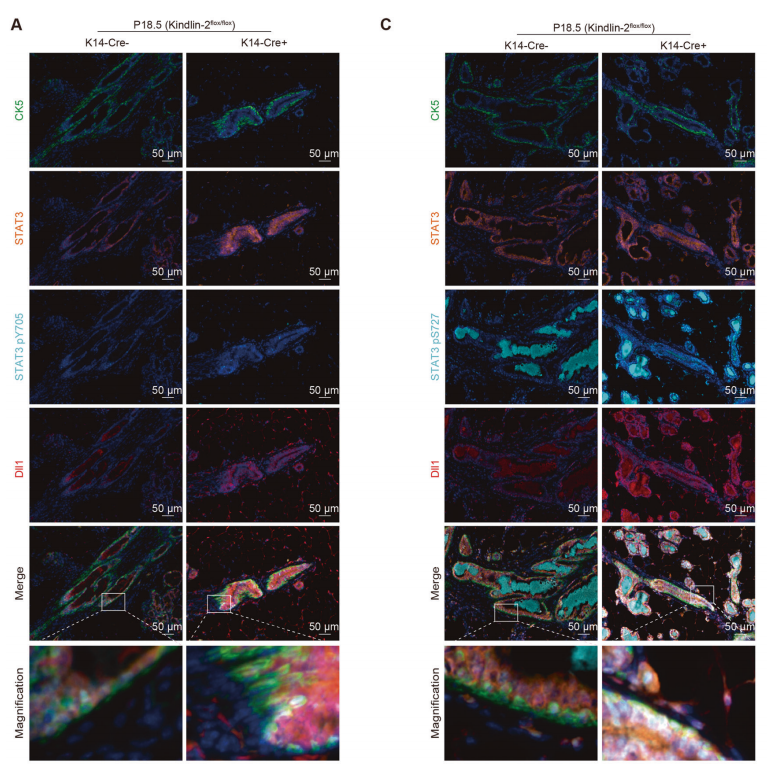
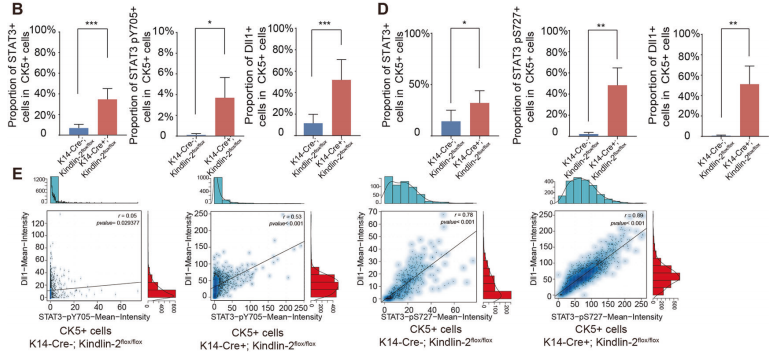
图6.Kindlin-2的缺失激活Stat3并上调肌上皮中的Dll1
7.桔皮素治疗可改善(K14-Cre+; Kindlin-2flox/flox)雌性小鼠的泌乳功能
肌上皮中Kindlin-2的缺失激活Stat3,并急剧增加肌上皮细胞和管腔祖细胞之间Notch通路的通讯。与广泛分布的磷酸化Stat3相比,Notch1在管腔和腺泡祖细胞中相对精确的表达提供了更好的潜在药物靶点。在本文中,作者假设用一种有效的Notch1抑制剂桔皮素(tangeretin)靶向Notch通路,可以挽救功能障碍并增强管腔上皮的分化。
实验结果表明,桔皮素治疗部分恢复了(K14-Cre+; Kindlin-2flox/flox)的泌乳能力(图7B)。四组乳腺组织学结果显示,桔皮素治疗后(K14-Cre+; Kindlin-2flox/flox)细胞的腺泡形成得到部分恢复(图7C-E)。免疫组化染色显示,桔皮素治疗后(K14-Cre+; Kindlin-2flox/flox)中Notch通路明显被抑制。
这些结果表明,桔皮素处理阻断了由肌上皮细胞Kindlin-2缺失引起的管腔细胞Notch通路的异常激活,支持了哺乳时管腔上皮的功能。
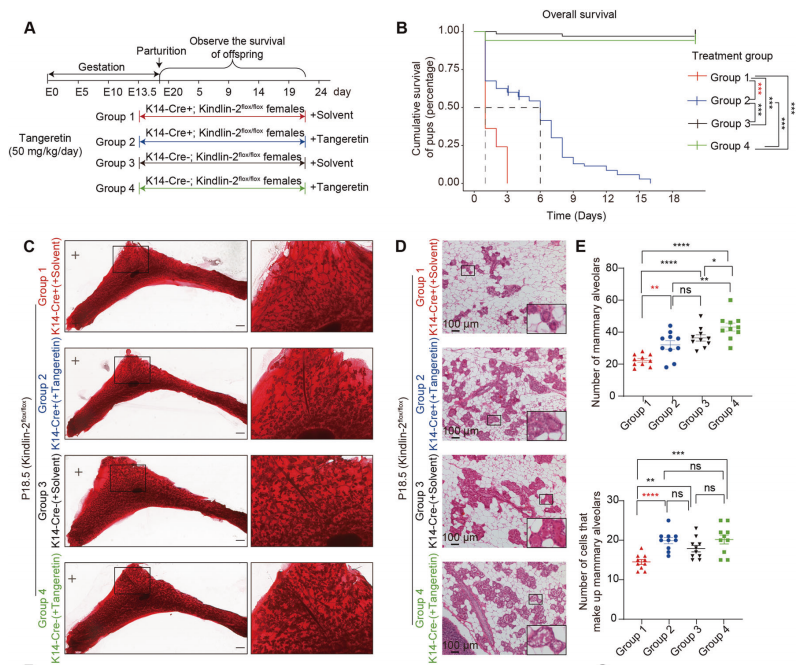
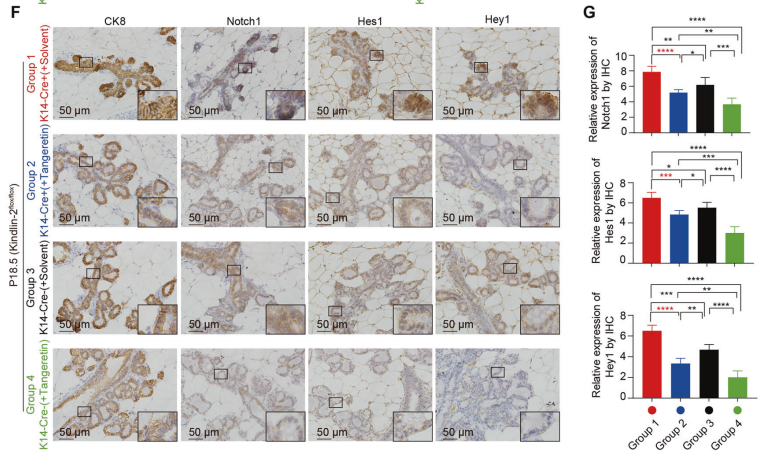
图7.桔皮素治疗可改善(K14-Cre+; Kindlin-2flox/flox)雌性小鼠的泌乳功能
结论
该研究揭示了Kindlin-2–Stat3–Dll1跨细胞调控乳腺发育异常和泌乳功能障碍的新分子机制。该研究在医疗卫生领域指导产后提升改善产妇哺乳能力,预防乳腺疾病以及乳制品制造业提升质量和产量中均具有重要的科学意义和潜在的转化价值。

参考文献
Wang Z, Zhang L, Li B, et al. Kindlin-2 in myoepithelium controls luminal progenitor commitment to alveoli in mouse mammary gland. Cell Death Dis. 2023 Oct 13;14(10):675. doi: 10.1038/s41419-023-06184-2. PMID: 37833248; PMCID: PMC10576046.







