脑电刺激作为前沿的脑机接口与控脑调节技术,正逐步成为神经调节与脑部疾病康复治疗领域的研究热点。特别是交流电刺激技术,通过其独特的交流电夹带效应(例如:40Hz的经颅交流电刺激)能够有效实现脑功能的精准调控。这一技术在调节脑电活动、提升认知能力、以及修复学习记忆障碍方面的应用,已经在近年的研究中得到了广泛报道。然而,关于交流电刺激对于脑部的局部与全局响应参数、特定响应神经细胞类群以及主要差异基因的影响等方面的研究,目前仍然依赖于更多的实验数据来进一步深化和验证。
项目文章 | 单细胞测序助力诱导全新类永生化功能性T细胞
发布时间:2024-05-29 10:33:32
2024年5月,清华大学基础医学院彭敏副教授团队在 Journal of Experimental Medicine 期刊(IF:11.743)发表了题为:Induction of immortal-like and functional CAR T cells by defined factors的研究成果。该研究报道了一种全新的T细胞状态——类永生化功能性T细胞(Immortal-like and Functional T cells,TIF)。TIF具有类似诱导多功能干细胞(iPSC)的自我更新能力,同时完整保留了成熟T细胞的生理功能,可用于开发更持久的CAR-T细胞,避免预后肿瘤复发。
新格元在该研究中提供了新格元GEXSCOPE®单细胞核转录组测序,用来分析CAR19TIF细胞群的异质性。
下面和元小新一起来看看吧~
研究背景
思维导图
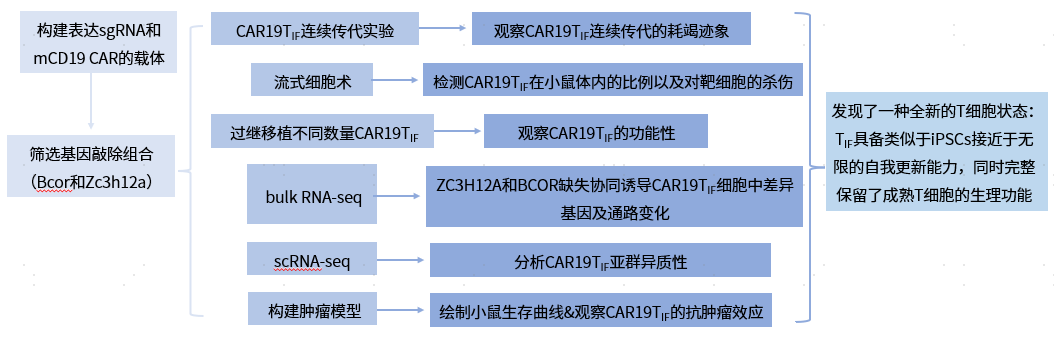
研究结果
1.通过抑制ZC3H12A和BCOR诱导永生化功能性CD19 CAR-T (CAR19TIF) 细胞
为了模拟CAR-T细胞在临床上识别并消灭B细胞的情况 ,作者使用了一种靶向小鼠CD19 (mCD19) 24的CD19 CAR细胞。在免疫功能正常的小鼠中,CAR19T细胞不扩增也不杀死CD19+细胞。为了解决这个问题,研究者筛选了一些基因,这些基因的缺失可以导致T细胞自发扩增,并且不影响其功能,其中Bcl2/11 /Fas、Ctla4、Tgfbr2和Zc3h12a基因位列首选。经过两轮筛选,作者确定了小鼠基因组中Zc3h12a和Bcor两个基因的抑制,产生了CAR19T细胞,这些细胞可以在免疫功能正常的小鼠体内扩增、持续存在并导致长期B细胞耗竭。并被命名为CAR19TIF。
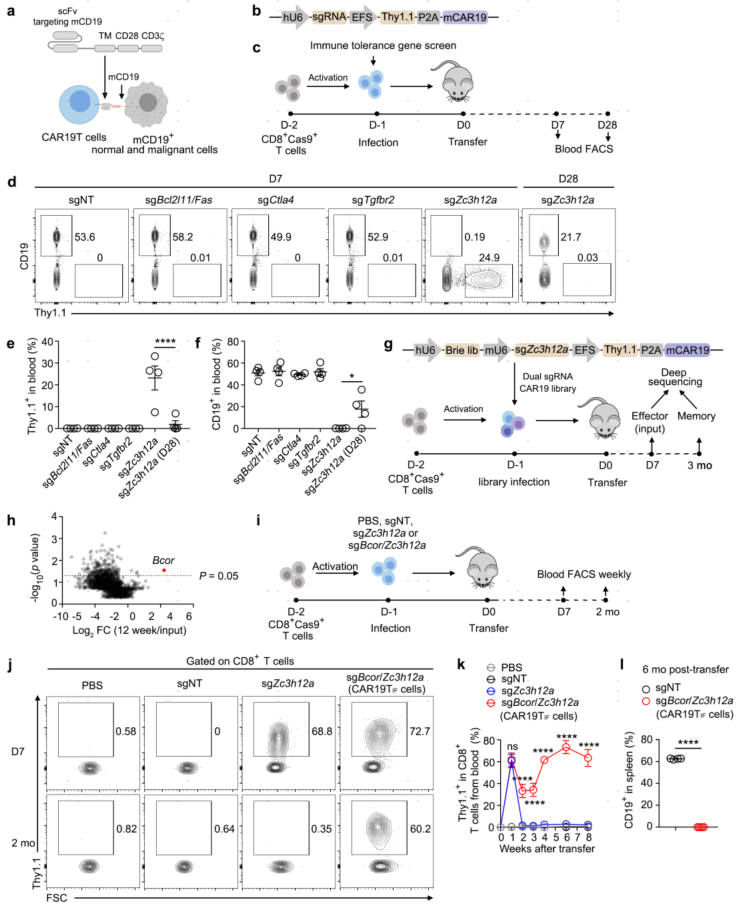
图1 敲除ZC3H12A和BCOR后产生类永生化功能性CAR19TIF细胞
2.CAR19TIF细胞兼具干性和功能性但无成瘤性
在检验哺乳动物细胞干性金标准的连续传代实验中,CAR19TIF在无需任何预处理的条件下可连续传代6次不表现出任何耗竭的迹象。类似的条件下造血干细胞(HSCs)仅能连续传代3~4次,因而CAR19TIF的干性远超HSCs,更接近iPSCs。以上数据表明CAR19TIF细胞像iPSCs一样具有无限的自我更新能力,且保留了成熟T细胞的功能。同时,CAR19TIF表现出了超强的功能性,500个CAR19TIF即可清除小鼠体内数以亿计的靶细胞。需要指出的是,CAR19TIF并没有转变为肿瘤细胞,其不能在体外存活,也不能在严重免疫缺陷的NSG小鼠中扩增和存活,不具备成瘤性。
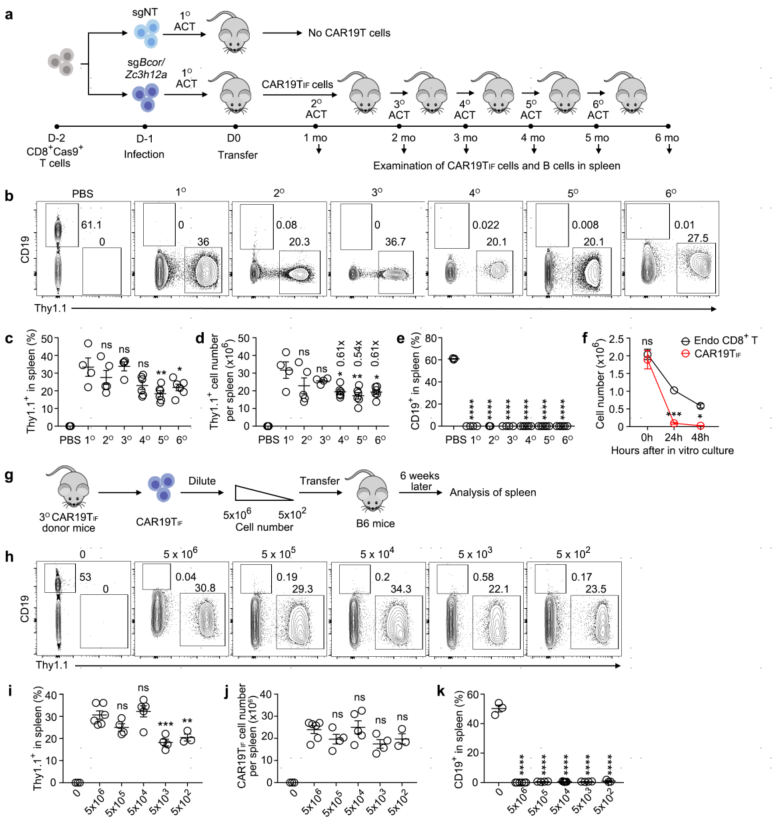
图2 CAR19TIF具有超强的功能性,且不具备成瘤性
3.CAR19TIF细胞表现出效应T细胞、记忆T细胞和前体耗尽T细胞的杂交特征
大部分CAR19TIF细胞小鼠的内源性脾CD8+ T细胞为CD62LhiCD44lo naïve T (TN)细胞,约10%为CD62LhiCD44hi TCM细胞。脾脏中约95%的CAR19TIF细胞为CD62LhiCD44hi,这是一种TCM细胞的表型。CAR19TIF细胞和内源性CD8+ T细胞中CD62L、TCF1、CD25、CD127和CD122的表达水平类似,再次提示CAR19TIF细胞具有TN或TCM细胞表型。CAR19TIF细胞还高表达了滤泡辅助性T细胞或TPEX细胞的标志物PD-1和CXCR5。CAR19TIF细胞也表达造血干细胞标志物c-Kit,但TSCM细胞的标志物CD150水平较低。PMA和离子霉素对CAR19TIF细胞的体外刺激显示,CAR19TIF细胞产生IL-2和IFNγ,与TCM细胞相似。体外杀伤实验显示,CAR19TIF细胞无需预扩增即可直接杀伤B细胞,证明这些细胞具有细胞毒性。这些数据表明,CAR19TIF细胞表现出独特的表型,不属于已知的T细胞亚群。
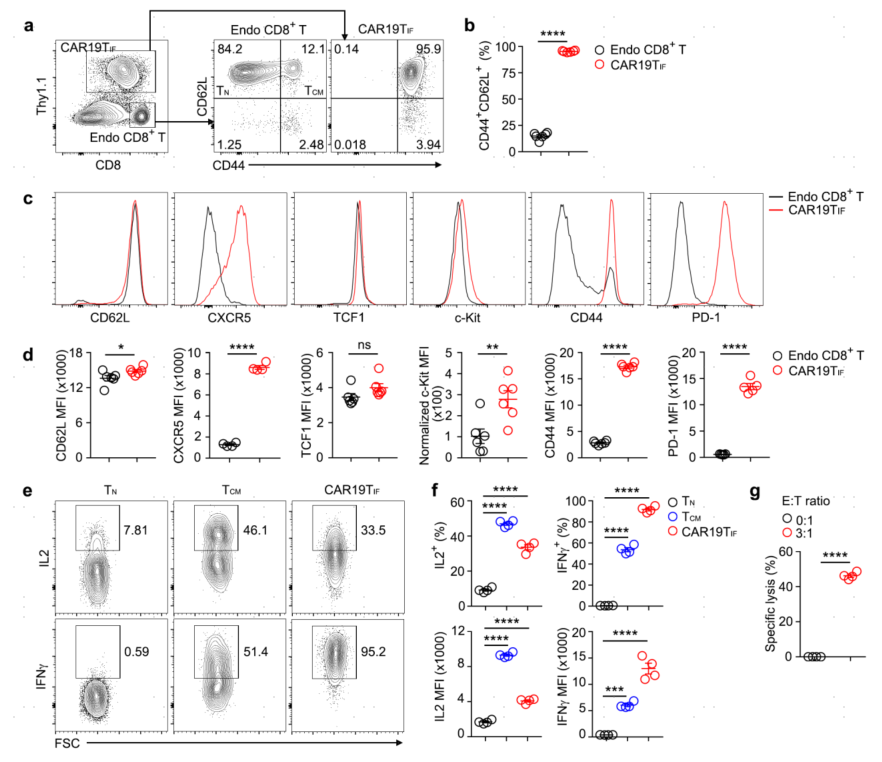
图3 CAR19TIF具有效应T细胞、记忆T细胞和前体耗竭T细胞的杂交特征
4.ZC3H12A和BCOR敲除协同重编码CAR19TIF细胞
为了探索CAR19TIF细胞重编程的机制,作者首先对脾脏分离的细胞进行了bulk RNA-seq分析。PCA分析显示,在转移后第10天,ZC3H12A敲除的CAR19T细胞收缩而ZC3H12A和BCOR双缺陷的CAR19TIF细胞持续存在,并且ZC3H12A缺陷的CAR19T细胞和CAR19TIF细胞的转录组也有明显差异。与单独缺乏ZC3H12A相比,CAR19TIF细胞中有1183个基因下调和239个基因上调。通路分析显示,CAR19TIF细胞中炎症相关模块被抑制,而多能性、干性和Wnt通路相关信号被上调,表明BCR缺失能够抑制炎症并诱导干性。
淋巴结归巢受体CCR7在CAR19TIF细胞中下调,证实了这些细胞在淋巴结中的低丰度。而CXCR4和CX3CR1等炎症趋化因子受体在CAR19TIF细胞中的表达增加,与它们的全身分布的情况一致。转录因子方面,与内源性CD8+ T细胞相比,CAR19TIF细胞中TCF1、TOX、256 TOX2、BATF、ZEB2、BLIMP1、BCL6和EOMES的表达增加,ID2和SATB1的表达下调。此外,CAR19TIF细胞还表达HOXB3和HOXB4。将CAR19TIF细胞的转录组与已知T细胞亚群的RNA-seq数据进行比较,发现CAR19TIF细胞具有干细胞样T细胞、TPEX细胞和效应T细胞的混合特征。通路分析显示,在CAR19TIF细胞中,Wnt信号高度富集,核糖体生物发生、翻译、MYC靶点和氧化磷酸化受到抑制,进一步证实了CAR19TIF细胞的干细胞特征和静止性。另外,单细胞数据表明CAR19TIF细胞的干性并不存在于特定的亚群中,并且干性基因和效应功能基因在同一细胞中的表达。综上所述,这些数据表明ZC3H12A-和BCOR -缺陷协同诱导CAR19TIF细胞中一个独特的程序,赋予这些细胞扩增能力、干细胞特性和静止性。
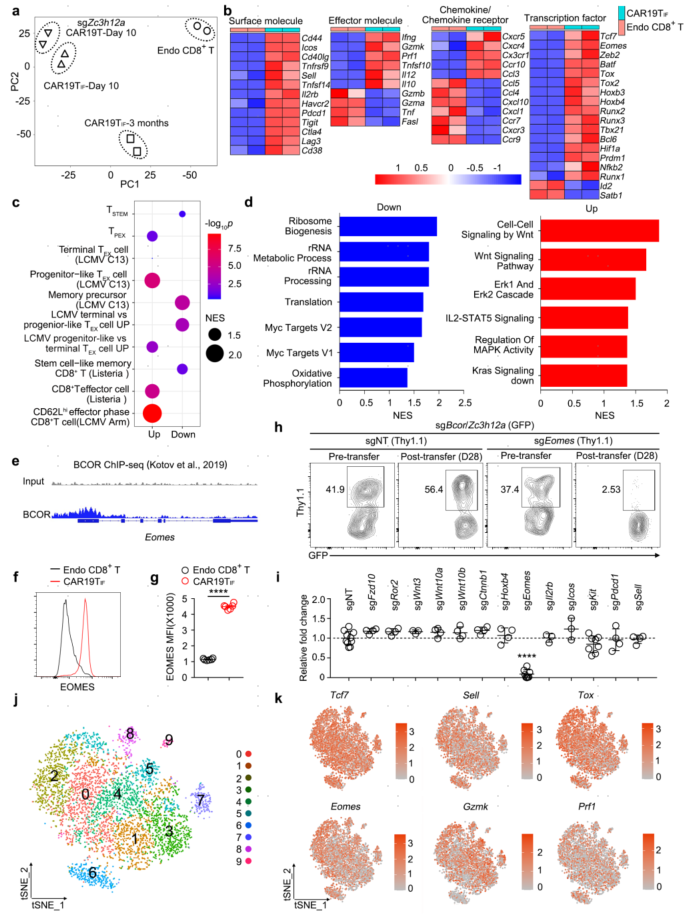
图4 ZC3H12A和BCOR同时敲除重新编码CAR19TIF细胞
5.CAR19TIF细胞具有长期抑制肿瘤的作用
CAR19TIF细胞在小鼠体内持续存在,这些小鼠在观察期间从未出现B细胞反弹。作者发现:CAR19TIF细胞持续监测和杀伤内源性和外源性CD19+细胞。CAR19TIF细胞抑制表达mCD19的MC38肿瘤细胞(MC38-mCD19)的生长,延长小鼠生存期。
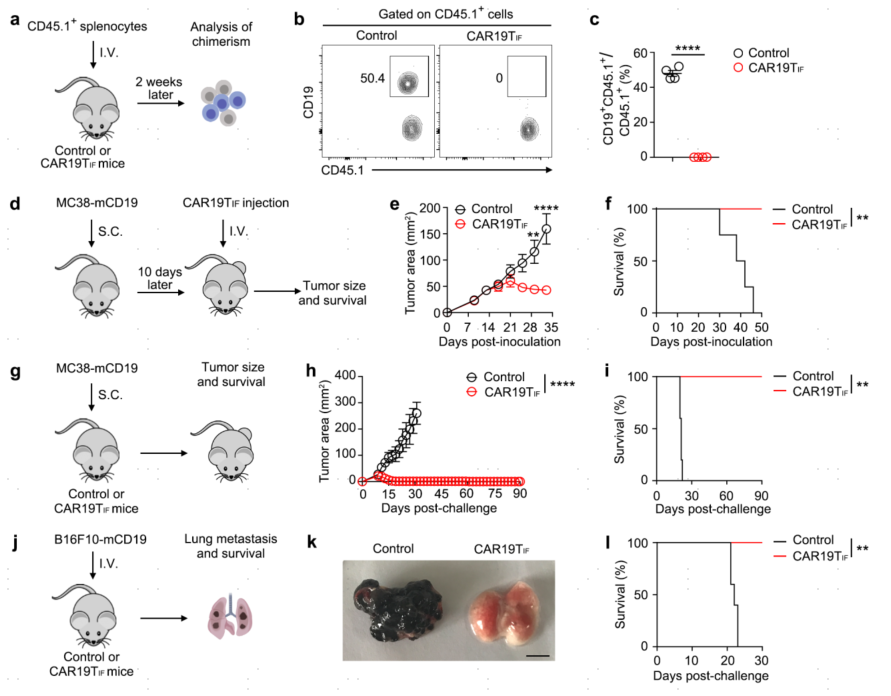
图5 CAR19TIF细胞具有长效抑制肿瘤作用
6.诱导带能靶向实体瘤抗原CARs的TIF细胞
作者首先测试了一种靶向人类EGFR的CAR,数据表明外周血中仅检测到表达靶向Bcor和Zc3h12a的sgRNAs的EGFR CAR-T细胞,在转移后4周达到峰值,并持续至少24周。ZC3H12A单独缺乏不能促进EGFR CAR-T细胞扩增,而BCOR和ZC3H12A双缺失可增强EGFR CAR-T细胞的扩增和持久性,因其与CAR19TIF细胞相似而被称为EGFRTIF细胞。EGFRTIF细胞可在B6或NSG小鼠中连续转移至少3代而不过度生长。这些数据表明EGFRTIF细胞具有优异的干性,但不会转化为癌细胞。
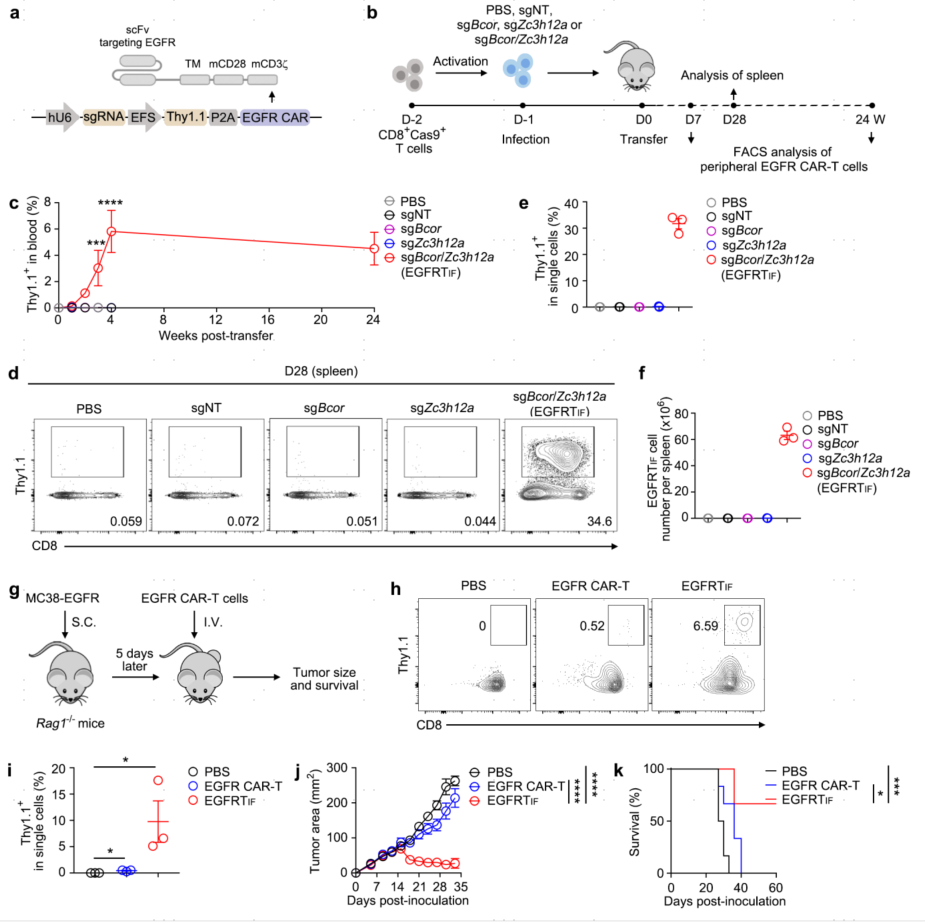
图6. EGFRTIF细胞诱导
7.GD2TIF细胞的诱导需要CAR tonic信号
在CD19 CAR缺乏大量靶细胞如正常B细胞的情况下,TIF程序的诱导可能依赖于CAR的tonic信号。为了验证这一假设,作者尝试了另一种具有少量tonic信号的CAR:来自4D5单克隆抗体的HER2 CAR。数据发现BCOR和ZC3H12A的缺失仅能最低限度地扩增HER2 CAR-T细胞。总而言之,GD2TIF细胞的诱导不需要抗原识别,因为来自GD2(14G2A) CAR或HER2 CAR的CDRs都可以工作,结合BCOR/ZC3H12A缺失,tonic信号足以在CAR靶细胞不可用时诱导TIF程序,如小鼠体内的HER2 CAR-T细胞。
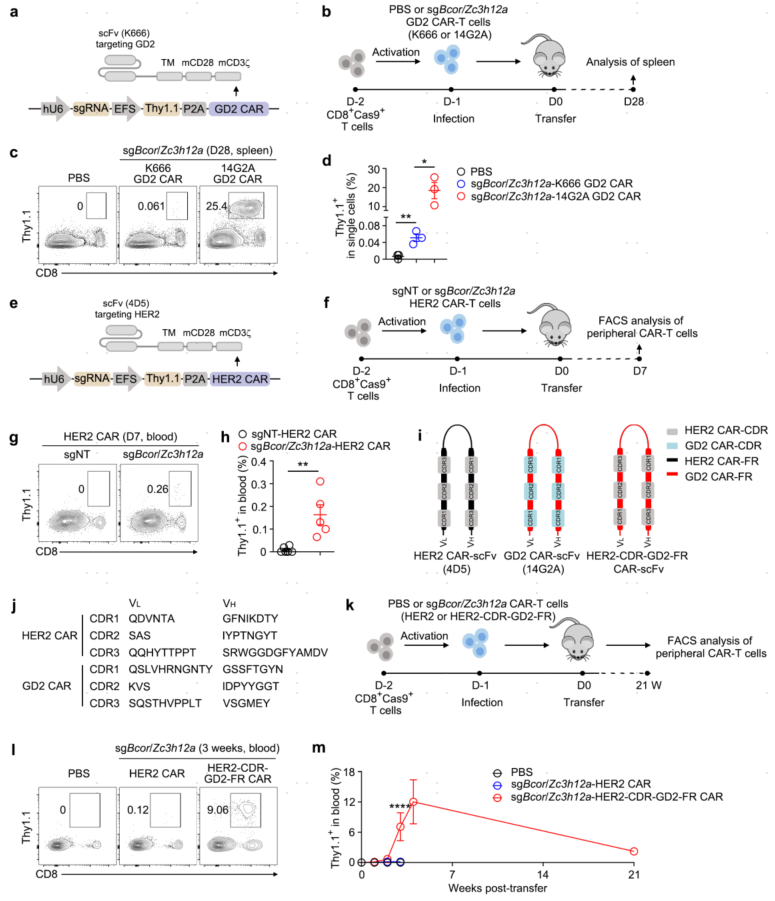
图7 GD2TIF细胞的诱导需要CAR tonic信号
8.人CAR-TIF细胞的诱导
最后,作者测试了TIF编码是否可以在人的T细胞中诱导。这些CAR表达(GFP+)和基因编辑的人T细胞被转移到NSG小鼠体内,以监测CAR-T细胞的扩增能力和持久性。这些BCOR/ZC3H12A双编辑的人GD2 CAR- T细胞在继发性NSG小鼠中可以扩增并持续存在,表明这些细胞具有良好的干性。以上数据表明:TIF也可在人的T细胞中诱导,并表现出和小鼠TIF细胞类似的特征。
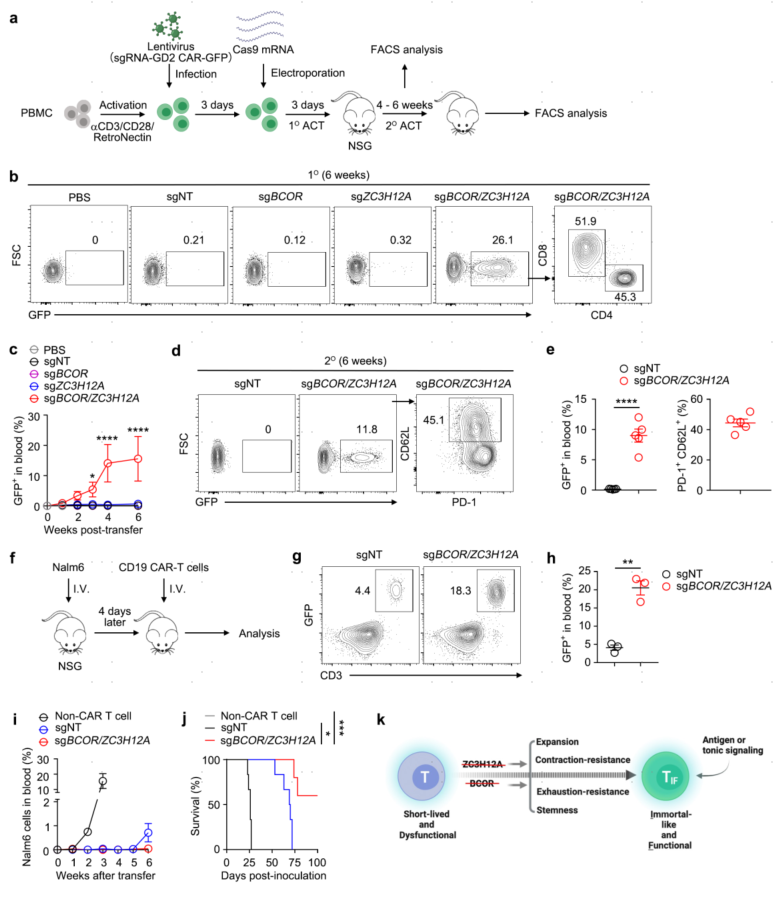
图8 人T细胞中CAR-TIF细胞诱导
参考文献
Wang L, Jin G, Zhou Q, et al. Induction of immortal-like and functional CAR T cells by defined factors. J Exp Med. 2024 May 6;221(5):e20232368. doi: 10.1084/jem.20232368.







