分析软件
DynaSCOPE®单细胞转录动态监测数据分析
一、简介
DynaSCOPE®单细胞转录动态监测试剂盒利用微流控系统,将mRNA代谢标记和高通量单细胞转录组测序整合,用cell barcode对数千至数万个细胞进行标记,不仅能够在单细胞层面检测到数千个基因的数量,同时检测到RNA转录本的“新”及“旧”,在丰富多维的单细胞测序信息进一步加上了时间的维度,实现更高通量的RNA转录动态检测。

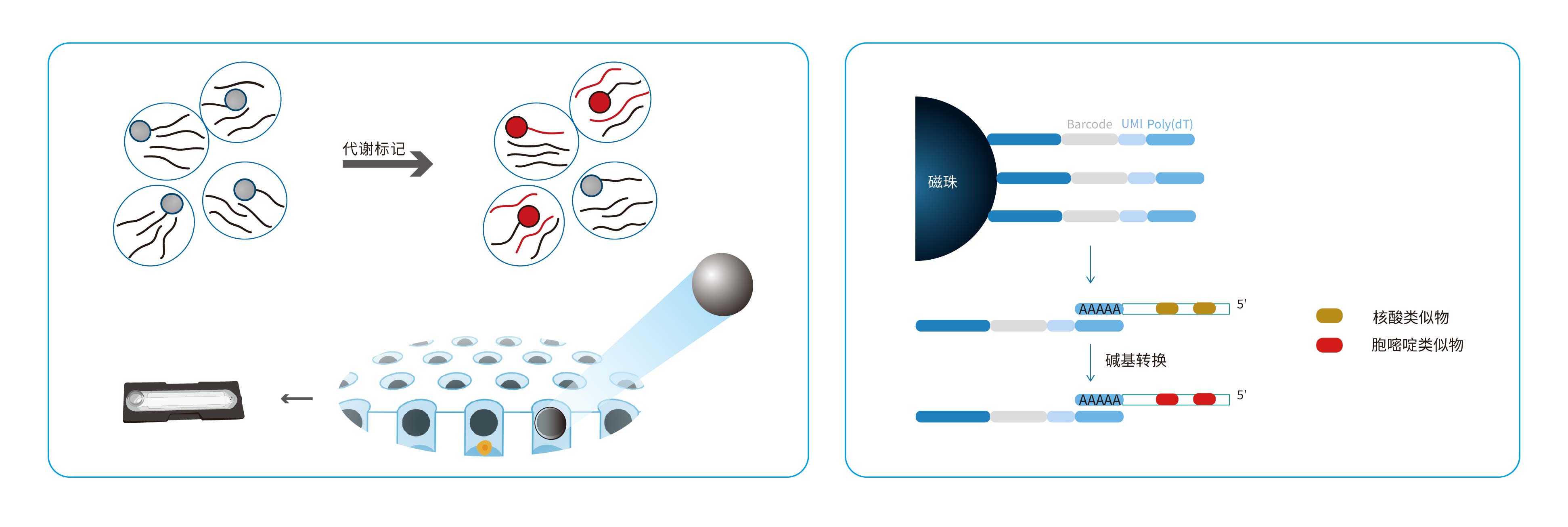
二、工作流程
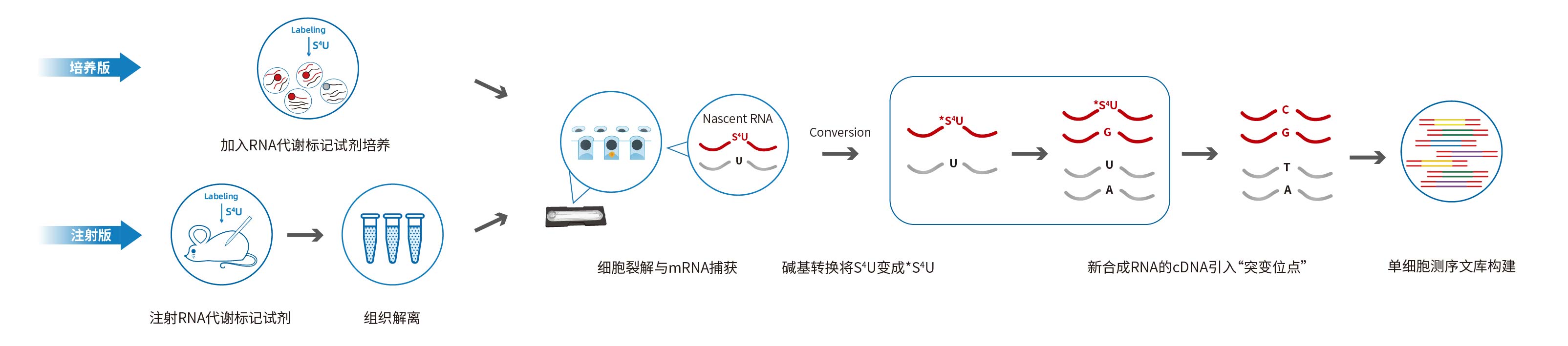
三、应用场景
- 转录组动力学研究——转录动态监测及调控
- 发育研究——发育及神经性机理研究
- 病毒感染研究——不同感染阶段机理研究
- 药物作用研究——药物潜在靶基因动态变化
四、celescope DynaScope 分析流程
可以通过 celescope dynaseq {指令} --help 查看:
$ celescope dynaseq --help
usage: celescope dynaseq [-h]
{sample,barcode,cutadapt,star,featureCounts,count,analysis,conversion,substitution,replacement,replace_tsne}
...
Single-cell dynaseq
positional arguments:
{sample,barcode,cutadapt,star,featureCounts,count,analysis,conversion,substitution,replacement,replace_tsne}
optional arguments:
-h, --help show this help message and exit
# 具体参数的详细说明,请参考GitHub:https://github.com/singleron-RD/CeleScope/blob/master/docs/dynaseq/multi_dynaseq.md下载测试数据与脚本
为了便于测试软件,我们在github上托管了测试数据(请注意,这些数据仅供测试用途。有些数据是人为生成的)。所有的软件DEMO测试数据我们已经在celescope``rna的教程中下载过,这里可以看一下vdj 数据的结构。
注意:Dynascope的DEMO测试数据是小鼠的,所以基因组需要构建对应物种的:Mus_ensembl_99。这里提供构建参考基因组所需的下载方式,构建命令与 Homo sapiens 一样。
mkdir test_dir
cd test_dir
wget ftp://ftp.ensembl.org/pub/release-99/fasta/mus_musculus/dna/Mus_musculus.GRCm38.dna.primary_assembly.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/gtf/mus_musculus/Mus_musculus.GRCm38.99.gtf.gz
gunzip Mus_musculus.GRCm38.dna.primary_assembly.fa.gz
gunzip Mus_musculus.GRCm38.99.gtf.gz
让我们看一看下载的测试数据
$ tree -L 2
.
|-- a1.vcf
|-- control.PosTag.csv
|-- dyna.mapfile
|-- fastqs
| |-- test_1.fq.gz
| `-- test_2.fq.gz
`-- gene.strandedness.csv
下面是测试脚本:
$ tree -L 2
.
|-- dyna.mapfile
`-- run_shell.sh
五、DynaSCOPE®单细胞转录动态监分析实操
介绍:DynaSCOPE®单细胞转录动态监测研究在实验过程中会构建一个单细胞转录组对照文库和一个DynaSCOPE®单细胞动态转录组文库,因此数据分析也就分为两个环节:
(1) 单细胞转录组背景文库测序
(2) DynaSCOPE®单细胞动态转录组文库测序
本篇文章内只介绍 DynaSCOPE®单细胞转录动态监分析 ,而celescope分析单细胞转录组数据的教程已在上一期中进行介绍。后续的分析流程和单细胞转录组分析保持一致的。
在分析之前我们先要激活我们celescope软件的运行环境,可以使用 conda activate celescope 命令进行激活。
(base) singleron 13:20:48 /dynaseq
$ conda activate celescope
(celescope) singleron 13:21:18 /dynaseq
然后就是接下来使用的两个重要的配置文件,run_shell.sh和dyna.mapfile
$ tree -L 1
.
|-- run_shell.sh
`-- dyna.mapfile
1、用 multi_dynaseq 构建 celescope dynaseq 分析的 shell 脚本
test .../celescope_test_data/dynaseq/fastqs/ dyna_test None .../celescope_test_data/dynaseq/a1.vcf
- 第一列:fastq 文件前缀(只写 _1.fq.gz 或 _2.fq.gz 前面的部分)
- 第二列:fastq 文件的路径(复制到文件夹即可)
- 第三列:对应样本名称(自己拟定)
- 第四列: (可选) 强制细胞数目,None 为不强制
- 第五列:背景 snp 文档
另一个是 shell 脚本文件:run_shell.sh
multi_dynaseq 必要参数
multi_dynaseq
--mapfile ../celescope_test/dynaseq/dyna.mapfile
--genomeDir ../ref_lib/mkref_datasets/mkref_Mus
--allowNoPolyT
--allowNoLinker
--STAR_param "--outFilterScoreMinOverLread 0.3 --outFilterMatchNminOverLread 0.3 --outSAMattributes MD"
--strand ../dynaseq/gene.strandedness.csv
--mod shell
- --mapfile mapfile文档的路径,写到文件名
- --genomeDir 参考基因组目录,写到文件夹
- --allowNoPolyT 接受没有 PolyT 的 valid reads,无值
- --allowNoLinke 接受没有正确 Linker 的 valid reads,无值
- --STAR_param star 软件的参数,按代码块内值进行设置(复制黏贴)
- --strand 基因组链的信息,提供文件路径,写到文件名
- --mod 生成脚本的格式
2、生成shell脚本
(1)运行刚编辑好的shell脚本run_shell.sh
$ sh run_shell.sh
(2)运行完以后就可以自动生成一个名称为shell的文件目录。
$ tree -L 1
.
|-- run_shell.sh
|-- shell
`-- dyna.mapfile
shell文件夹中会有一个以dyna_test命名的脚本运行存储数据的目录,以及一个运行的shell脚本dyna_test.sh,dyna_test.sh脚本中的每行指令对应每一步分析(质控报告的每一部分数据)。
3、投递shell脚本
$ tree -L 1
.
|-- dyna_test
|-- dyna_test.sh
`-- nohup.out
如果对每一步做了什么感兴趣,可以单独运行查看,dyna_test.sh 里面是:
$ cat test1.sh
celescope dynaseq sample --outdir .//test1/00.sample --sample test1 --assay dynaseq --thread 4 --chemistry auto --fq1 /mnt/sdd/singleron_training_class/resources/celescope_test/dynaseq/fastqs/test_1.fq.gz
celescope dynaseq barcode --outdir .//test1/01.barcode --sample test1 --assay dynaseq --thread 4 --chemistry auto --lowNum 2 --allowNoPolyT --allowNoLinker --fq1 /mnt/sdd/singleron_training_class/resources/celescope_test/dynaseq/fastqs/test_1.fq.gz --fq2 /mnt/sdd/singleron_training_class/resources/celescope_test/dynaseq/fastqs/test_2.fq.gz
celescope dynaseq cutadapt --outdir .//test1/02.cutadapt --sample test1 --assay dynaseq --thread 4 --minimum_length 20 --nextseq_trim 20 --overlap 10 --insert 150 --fq .//test1/01.barcode/test1_2.fq
celescope dynaseq star --outdir .//test1/03.star --sample test1 --assay dynaseq --thread 4 --genomeDir /mnt/sdd/singleron_training_class/resources/ref_lib/mkref_datasets/mkref_Mus --STAR_param "--outFilterScoreMinOverLread 0.3 --outFilterMatchNminOverLread 0.3 --outSAMattributes MD" --outFilterMultimapNmax 1 --starMem 30 --fq .//test1/02.cutadapt/test1_clean_2.fq
celescope dynaseq featureCounts --outdir .//test1/04.featureCounts --sample test1 --assay dynaseq --thread 4 --gtf_type exon --genomeDir /mnt/sdd/singleron_training_class/resources/ref_lib/mkref_datasets/mkref_Mus --input .//test1/03.star/test1_Aligned.sortedByCoord.out.bam
celescope dynaseq count --outdir .//test1/05.count --sample test1 --assay dynaseq --thread 4 --genomeDir /mnt/sdd/singleron_training_class/resources/ref_lib/mkref_datasets/mkref_Mus --expected_cell_num 3000 --cell_calling_method auto --bam .//test1/04.featureCounts/test1_name_sorted.bam --force_cell_num None
celescope dynaseq analysis --outdir .//test1/06.analysis --sample test1 --assay dynaseq --thread 4 --genomeDir /mnt/sdd/singleron_training_class/resources/ref_lib/mkref_datasets/mkref_Mus --matrix_file .//test1/05.count/test1_matrix_10X
celescope dynaseq conversion --outdir .//test1/07.conversion --sample test1 --assay dynaseq --thread 4 --strand /mnt/sdd/singleron_training_class/resources/celescope_test/dynaseq/gene.strandedness.csv --bam .//test1/04.featureCounts/test1_Aligned.sortedByCoord.out.bam.featureCounts.bam --cell .//test1/05.count/test1_matrix_10X/barcodes.tsv
celescope dynaseq substitution --outdir .//test1/08.substitution --sample test1 --assay dynaseq --thread 4 --bam .//test1/07.conversion/test1.PosTag.bam
celescope dynaseq replacement --outdir .//test1/09.replacement --sample test1 --assay dynaseq --thread 4 --bg_cov 1 --bam .//test1/07.conversion/test1.PosTag.bam --bg /mnt/sdd/singleron_training_class/resources/celescope_test/dynaseq/a1.vcf
celescope dynaseq replace_tsne --outdir .//test1/10.replace_tsne --sample test1 --assay dynaseq --thread 4 --tsne .//test1/06.analysis/test1_tsne_coord.tsv --mat .//test1/09.replacement/test1.fraction_of_newRNA_matrix.txt --rep .//test1/09.replacement/test1.fraction_of_newRNA_per_cell.txt
4、结果目录
运行完成后,可以看一下结果文件,其目录如下:
$ tree ./
./
|-- 00.sample
| `-- stat.txt
|-- 01.barcode
| |-- stat.txt
| `-- test1_2.fq
|-- 02.cutadapt
| |-- cutadapt.log
| |-- stat.txt
| `-- test1_clean_2.fq
|-- 03.star
| |-- stat.txt
| |-- test1_Aligned.out.bam
| |-- test1_Aligned.sortedByCoord.out.bam
| |-- test1_Aligned.sortedByCoord.out.bam.bai
| |-- test1_Log.final.out
| |-- test1_Log.out
| |-- test1_Log.progress.out
| |-- test1_SJ.out.tab
| `-- test1_region.log
|-- 04.featureCounts
| |-- stat.txt
| |-- test1
| |-- test1.summary
| `-- test1_name_sorted.bam
|-- 05.count
| |-- stat.txt
| |-- test1_all_matrix
| | |-- barcodes.tsv
| | |-- genes.tsv
| | `-- matrix.mtx
| |-- test1_count_detail.txt
| |-- test1_counts.txt
| |-- test1_downsample.txt
| `-- test1_matrix_10X
| |-- barcodes.tsv
| |-- genes.tsv
| `-- matrix.mtx
|-- 06.analysis
| |-- mito.txt
| |-- stat.txt
| |-- test1_markers.tsv
| `-- test1_tsne_coord.tsv
|-- 07.conversion
| |-- stat.txt
| |-- test1.PosTag.bam
| |-- test1.PosTag.bam.bai
| `-- test1.PosTag.csv
|-- 08.substitution
| |-- stat.txt
| `-- test1.substitution.txt
|-- 09.replacement
| |-- stat.txt
| |-- test1.TC_matrix.rds
| |-- test1.fraction_of_newRNA_matrix.txt
| |-- test1.fraction_of_newRNA_per_cell.txt
| |-- test1.fraction_of_newRNA_per_gene.txt
| |-- test1.new_matrix.tsv.gz
| `-- test1.old_matrix.tsv.gz
|-- 10.replace_tsne
| |-- stat.txt
| |-- test1.rep_in_tsne.txt
| `-- test1.rep_in_tsne_top10.txt
`-- test1_report.html
13 directories, 51 files
当运行完结束,就可以得到一个单细胞动态检测转录组数据的网页版标准报告。质控报告的样本和软件的基本信息:
- 基因组比对信息
- 生成表达矩阵
- 降维聚类分析
- 动态转录分析
- 所有reads的总转换率
- 新生转录本的表达矩阵
- 簇中的高转换基因列表
后续脚本生成和运行步骤和转录组一样,几个重要文件的提取路径如下:
所有reads的总转换率:【08.substitution】 内 {sample}.substitution.txt
新生转录本的表达矩阵 :【09.replacement】内 {sample}.new_matrix.tsv.gz
tsNE分群和差异基因信息:【10.replace_tsne】
如需更多参数设定,请参考Github说明文档
CeleScope/multi_dynaseq.md at master · singleron-RD/CeleScope (github.com)
好啦,以上就是一个完整的新格元DynaSCOPE™单细胞转录动态监测试剂盒分析流程,接下来就可以进行数据分析了。
本文仅做软件安装测试使用,更多软件更新信息参见:https://github.com/singleron-RD/CeleScope







