分析软件
AccuraCode®高通量药物筛选RNA数据分析
一、简介
AccuraCode 是一款高通量药物筛选试剂盒,可一次完成数百种药物处理细胞的RNA捕获及混合建库,能快速获得对每种药物处理后细胞的基因表达信息,经生信数据分析,可获取到转录组药物评价信息,进而加速目标药物的筛选进程。 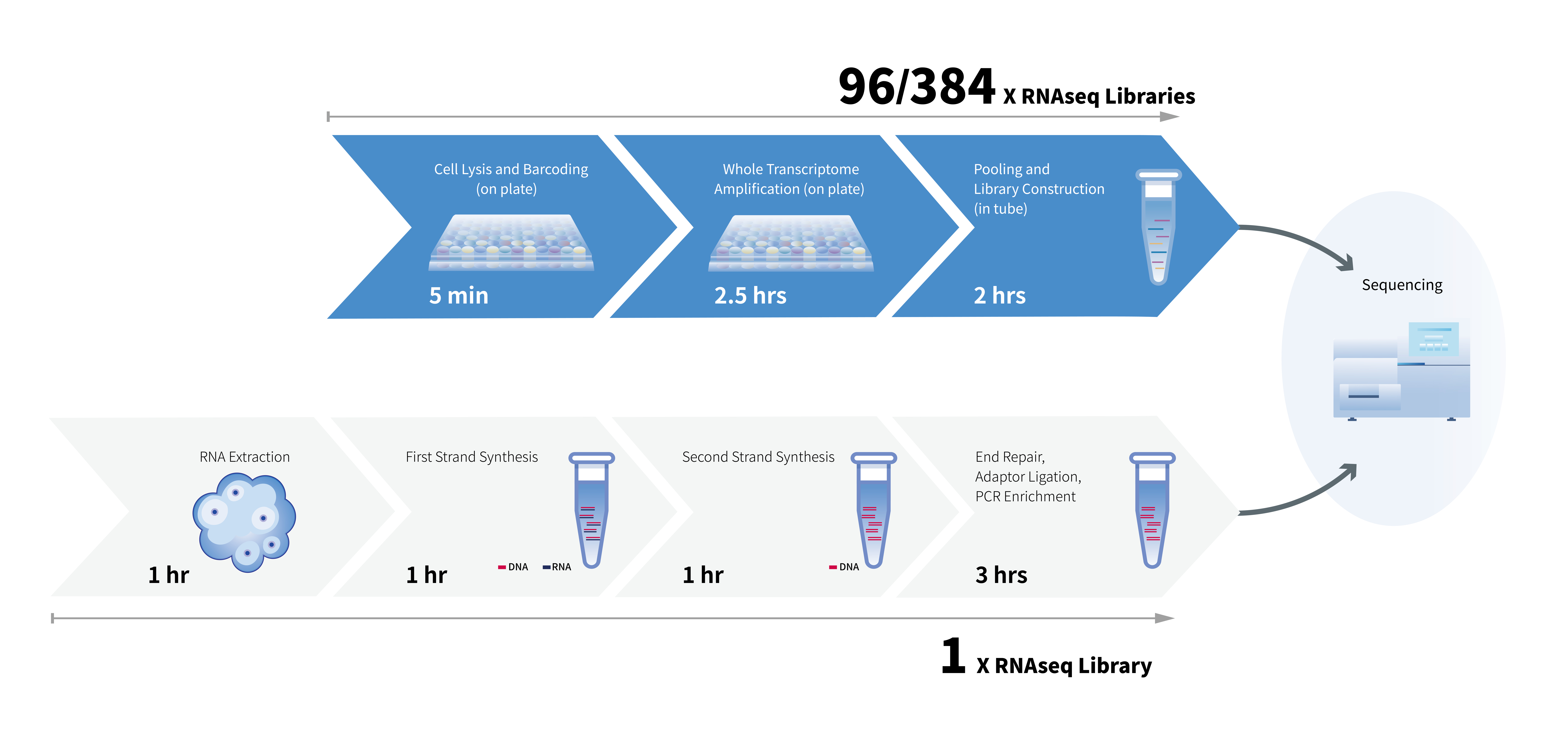
试剂盒结合了样本条形码 (Well Barcode) 和一步法逆转录扩增反应(OneStep WTA),高效捕获各孔中cDNA分子,然后混合纯化,可构建一个含多种药物细胞处理信息的转录组文库,测序后根据Well Barcode信息拆分出不同药物处理的转录本信息。 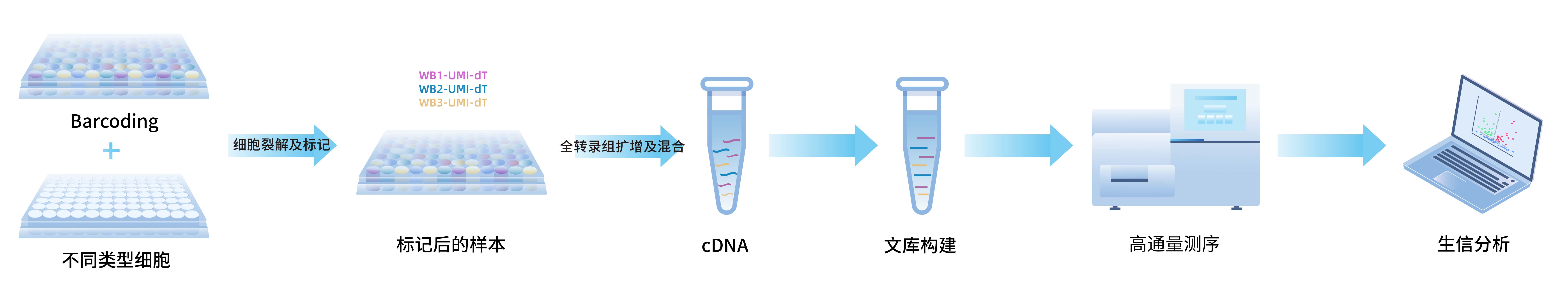
新格元的 accuracode软件 提供了接口程序(interfaces)multi_accura来生成流程脚本。
二、产品优势
药物筛选时间更节省— 将反转录及扩增两个反应合并为一步wTA,简化实验操作的同时,可节约实验时间适用多种细胞类型— 对贴壁细胞和悬浮细胞适用试剂组分全面—试剂盒包含从细胞裂解、RNA捕获、反转录到文库构建的全流程试剂搭配自动化通量更高—可搭配自动化工作站,实现批量筛选配备生信分析软件—提供基础版生信数据分析软件,可实现对数据的自主分析获取信息更全面—基于药物处理细胞中转录组变化信息,一次实验即可获取多基因信息
三、accuracode软件安装
accuracode 配置要求
- accuracode 硬件需求:Linux服务器运行内存推荐64G,最小内存32G, 硬盘空间建议大于1TB.
- accuracode 环境:conda (用于安装软件和环境管理),git (克隆 accuracode 源代码).
- 在输入命令前先了解输出结果,规范目录管理并做好记录.
- 在安装完软件之后先DEMO用测试rna数据跑一遍,保证依赖的环境均配置好
测试数据与脚本 软件仓库:https://github.com/singleron-RD/AccuraCode
药筛:WellBarcode:https://github.com/singleron-RD/AccuraCode_WellBarcode
测试数据与脚本:https://github.com/singleron-RD/AccuraCode_test_data
accuracode 环境配置
1. 克隆git上的软件库
$ git clone https://github.com/singleron-RD/AccuraCode.git
clone完成之后,看一下我们AccuraCode源码结构:
$ tree -L 1
.
|-- LICENSE
|-- MANIFEST.in
|-- README.md
|-- accuracode
|-- accuracode.egg-info
|-- accuracode.yml
|-- accuracode_log.txt
|-- build
|-- conda_pkgs.txt
|-- dist
|-- docs
|-- requirements.txt
`-- setup.py
2. 用conda 创建accuracode环境
# -n 后面是环境的名称,您可以取其他喜欢的名字
$ conda create -n accuracode -y --file conda_pkgs.txt # 最后这个文件里面是环境需要的软件
如果嫌 conda 太慢可以用 mamba 来提高速度,mamba也可以用来创建conda环境。
3. 安装conda包
cd AccuraCode
conda create -n accuracode
conda activate accuracode
conda install -y --file conda_pkgs.txt --channel conda-forge --channel bioconda --channel r --channel imperial-college-research-computing
4. 安装accuracode
在用 pip install accuracode 安装之前,请确认已经激活刚才创建的accuracode的环境.
$ conda activate accuracode
$ pip install accuracode
如果这里的pip install accuracode过程比较慢,也可以加上对应的镜像:
# 在中国可以给pip加上镜像来提速
$ which pip # check pip version and environment
$ pip install accuracode --default-timeout=100 -i https://pypi.tuna.tsinghua.edu.cn/simple # You can try other mirrors
安装过程中如果连接超时,可以在搜索频道后用 conda install 单独安装(scipy,numpy等软件容易断链)
python setup.py install
5. 检查安装版本
软件安装完成之后,可以检查一下:
which accuracode
accuracode --help
可以看到帮助信息:
usage: accuracode [-h] [-v] {accura} ...
AccuraCode
positional arguments:
{accura}
optional arguments:
-h, --help show this help message and exit
-v, --version show program's version number and exit
查看安装的accuracode的版本
$ accuracode -v
1.0.0 # 1.0.0是我们的使用版本
下次运行accuracode 之前只需要用conda activate accuracode来激活软件环境就可以使用accuracode来分析新格元单细胞数据啦。
在软件安装好后输入 multi_accura检查安装是否成功并获得帮助。
四、accuracode分析流程
accuracode生成的高通量药物筛选数据分析过程的基本流程。
accuracode pipeline (单细胞高通量药物筛选数据分析) 包含七个主要指令,可以通过accuracode accura{指令} --help 查看:
$ accuracode accura --help
usage: accuracode accura [-h]
{mkref,sample,barcode,cutadapt,star,featureCounts,count}
...
AccuraCode
positional arguments:
{mkref,sample,barcode,cutadapt,star,featureCounts,count}
optional arguments:
-h, --help show this help message and exit1. mkref 指令创建一个参考基因组文件
mkref 用于创建基因组参考目录,产生文件在当前工作目录下。每个物种构建一次参考基因组即可,以后运行直接读取,不用多次构建。
需要基因组序列文件与注释信息:fasta 文件和 gtf 文件
- 下面我们以Homo sapiens为例来演示基因组构建过程。
下载基因组序列文件与注释信息
mkdir hs_ensembl_99
cd hs_ensembl_99
wget ftp://ftp.ensembl.org/pub/release-99/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
wget ftp://ftp.ensembl.org/pub/release-99/gtf/homo_sapiens/Homo_sapiens.GRCh38.99.gtf.gz
gunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
gunzip Homo_sapiens.GRCh38.99.gtf.gz
2. 利用 accura mkref 构建参考基因组目录
注意:accura mkref 构建好的参考基因组目录需要包含下载的 fa 和 gtf 文件
conda activate accuracode
accuracode accura mkref \
--genome_name Homo_sapiens_ensembl_99 \
--fasta Homo_sapiens.GRCh38.dna.primary_assembly.fa \
--gtf Homo_sapiens.GRCh38.99.gtf
3. 构建好的参考基因组目录结构如下:
$ tree Homo_ref/
Homo_ref/
|-- Genome
|-- Homo_sapiens.GRCh38.99.gtf # gtf文件不是accur mkref生成的,但需要和生成文件在同一目录下
|-- Homo_sapiens.GRCh38.dna.primary_assembly.fa # fa文件不是accur mkref生成的,但需要和生成文件在同一目录下
|-- Homo_sapiens_ensembl_99.refFlat
|-- Log.out
|-- SA
|-- SAindex
|-- accuracode_genome.config
|-- accuracode_log.txt
|-- chrLength.txt
|-- chrName.txt
|-- chrNameLength.txt
|-- chrStart.txt
|-- exonGeTrInfo.tab
|-- exonInfo.tab
|-- geneInfo.tab
|-- genomeParameters.txt
|-- sjdbInfo.txt
|-- sjdbList.fromGTF.out.tab
|-- sjdbList.out.tab
`-- transcriptInfo.tab
4. 下载测试数据与脚本
为了便于测试软件,我们在github上托管了测试数据(请注意,这些数据仅供测试用途。有些数据是人为生成的)。
mkdir test_dir
cd test_dir
git clone https://github.com/singleron-RD/AccuraCode_test_data
当然,也可以在gitee上下载:
让我们看一看下载的测试数据和脚本
$ tree -L 2
.
|-- 384WellBarcode
|-- 96WellBarcode
|-- bclist
|-- fastqs
| |-- test_R1.fq.gz
| `-- test_R2.fq.gz
|-- mapfile
`-- multi_accura.sh
五、AccuraCode®高通量药物筛选分析实操
AccuraCode®高通量药物筛选分析研究在实验过程中也会构建一个转录组文库整体的分析和我们之前介绍的 multi_rna 基本一致,只需要增加 wellbarcode 的序列文件。
在分析之前我们先要激活我们accuracode软件的运行环境,可以使用conda activate accuracode命令进行激活。
(base) singleron 15:20:48 ../AccuraCode
$ conda activate accuracode
(accuracode) singleron 13:21:18 ../AccuraCode
然后就是接下来要使用的两个重要的配置文件,accura.mapfile和multi_accura.sh
$ tree -L 1
.
|-- accura.mapfile
`-- multi_accura.sh
1、用 multi_accura构建accuracode分析的 shell 脚本
配置 mapfile文件
--mapfile 是multi_accura下的参数,需要提供一个制表符分隔 (tab-delimited) 的文本文件。mapfile 的每一行代表双端 (paired-end) fastq文件。
test .../test_project/fastqs/ AccuraTest
第一列:fastq文件的前缀。注意只保留fastq文件名 _R1.fq .gz和 _R2.fq.gz 前的内容,如文件 test_R1.fq.gz 的前缀就是 test
第二列:fastq文件的保存位置,写到上一级目录,不用写到 fastq 文件
第三列:样本名(自拟),会成为后续脚本的前缀
另一个是 shell 脚本文件:multi_accura.sh
multi_accura\
--mapfile /mnt/sdd/singleron_training_class/resources/accuracode_project/test_project\
--genomeDir /mnt/sdd/singleron_training_class/resources/accuracode_project/test_project/Homo_ref\
--thread 8\
--mod shell\
--whitelist /mnt/sdd/singleron_training_class/resources/accuracode_project/test_project/bclist
- 第一行 --mapfile:之前配置好的mapfile的保存路径,写到mapfile文件
- 第二行 --genomeDir:构建好的参考基因组目录的路径,写到上一级目录
- 第三行 --thread:使用线程数
- 第四行 --mod:生成脚本的形式
- 第五行 --whitelist:wellbarcode 文档的保存路径,写到 wellbarcode 文件
2、生成shell脚本
(1)运行刚编辑好的shell脚本multi_accura.sh
$ sh multi_accura.sh
(2)运行完以后就可以自动生成一个名称为shell的文件目录。
$ tree -L 1
.
|-- multi_accura.sh
|-- shell
`-- accura.mapfile
shell文件夹中会有一个以accura_test命名的脚本运行存储数据的目录,以及一个运行的shell脚本accura_test.sh,accura_test.sh脚本中的每行指令对应每一步分析(质控报告的每一部分数据)。
3、投递shell脚本
进入到shell目录中,就可以运行shell脚本accura_test.sh,然后在终端命令行中输入nohup sh accura_test.sh &。将脚本提交到后台运行,具体细节已在上一期中详细介绍过了,就不过多赘述了。
$ tree -L 1
.
|-- accura_test
|-- accura_test.sh
`-- nohup.out
如果对每一步做了什么感兴趣,可以单独运行查看,accura_test.sh 里面是:
$ cat accura_test.sh
accuracode accura sample --outdir .//AccuraTest/00.sample --sample AccuraTest --assay accura --thread 8 --chemistry accuracode384 --fq1 /mnt/sdd/singleron_training_class/resources/accuracode_project/test_project/fastqs/test_R1.fq.gz
accuracode accura barcode --outdir .//AccuraTest/01.barcode --sample AccuraTest --assay accura --thread 8 --chemistry accuracode384 --whitelist /mnt/sdd/singleron_training_class/resources/accuracode_project/test_project/bclist --lowNum 2 --fq1 /mnt/sdd/singleron_training_class/resources/accuracode_project/test_project/fastqs/test_R1.fq.gz --fq2 /mnt/sdd/singleron_training_class/resources/accuracode_project/test_project/fastqs/test_R2.fq.gz
accuracode accura cutadapt --outdir .//AccuraTest/02.cutadapt --sample AccuraTest --assay accura --thread 8 --minimum_length 20 --nextseq_trim 20 --overlap 10 --insert 150 --fq .//AccuraTest/01.barcode/AccuraTest_2.fq
accuracode accura star --outdir .//AccuraTest/03.star --sample AccuraTest --assay accura --thread 8 --genomeDir /mnt/sdd/singleron_training_class/resources/accuracode_project/test_project/Homo_ref --outFilterMultimapNmax 1 --starMem 30 --fq .//AccuraTest/02.cutadapt/AccuraTest_clean_2.fq
accuracode accura featureCounts --outdir .//AccuraTest/04.featureCounts --sample AccuraTest --assay accura --thread 8 --gtf_type exon --genomeDir /mnt/sdd/singleron_training_class/resources/accuracode_project/test_project/Homo_ref --input .//AccuraTest/03.star/AccuraTest_Aligned.sortedByCoord.out.bam
accuracode accura count --outdir .//AccuraTest/05.count --sample AccuraTest --assay accura --thread 8 --genomeDir /mnt/sdd/singleron_training_class/resources/accuracode_project/test_project/Homo_ref --chemistry accuracode384 --whitelist /mnt/sdd/singleron_training_class/resources/accuracode_project/test_project/bclist --bam .//AccuraTest/04.featureCounts/AccuraTest_name_sorted.bam
4、结果目录
运行完成后,可以看一下结果文件,其目录如下:
$ tree accura_test
accura_test
|-- 00.sample
| `-- stat.txt
|-- 01.barcode
| |-- AccuraTest_2.fq
| `-- stat.txt
|-- 02.cutadapt
| |-- AccuraTest_clean_2.fq
| |-- cutadapt.log
| `-- stat.txt
|-- 03.star
| |-- AccuraTest_Aligned.out.bam
| |-- AccuraTest_Aligned.sortedByCoord.out.bam
| |-- AccuraTest_Aligned.sortedByCoord.out.bam.bai
| |-- AccuraTest_Log.final.out
| |-- AccuraTest_Log.out
| |-- AccuraTest_Log.progress.out
| |-- AccuraTest_SJ.out.tab
| |-- AccuraTest_region.log
| `-- stat.txt
|-- 04.featureCounts
| |-- AccuraTest
| |-- AccuraTest.summary
| |-- AccuraTest_Aligned.sortedByCoord.out.bam.featureCounts.bam
| |-- AccuraTest_name_sorted.bam
| `-- stat.txt
|-- 05.count
| |-- AccuraTest_count_detail.txt
| |-- AccuraTest_counts.txt
| |-- AccuraTest_downsample.txt
| |-- AccuraTest_matrix.tsv.gz
| `-- stat.txt
`-- accura_test_report.html
当运行完结束,就可以得到一个单细胞多样同测数据的网页版标准报告。
-
质控报告的样本和软件的基本信息 -
数据质控信息 -
生成表达矩阵 -
降维聚类分析 -
样本标签拆分后的细胞基因表达矩阵
附录:wellbarcode列表
well barcode 序列信息由新格元提供,可以自己用文本编辑器编辑一个,一行一个barcode,形式如下:
AACGGACCT
GACTGGTTG
TCGGTTCGT
GTCTTGCGT
具体的wellbarcode信息可以上git下载:
singleron-RD/AccuraCode_WellBarcode: Well Barcode sequences for AccuraCode® (github.com)
好啦,以上就是一个完整的新格元AccuraCode®高通量药物筛选分析过程,接下来就可以进行数据分析了。另外透漏一下,得到的表达矩阵是可以上传到新格元单细胞临床数据库SynEcoSys(https://www.synecosys.cn/)进行细胞注释和分析的哟~
本文仅做软件安装测试使用,更多软件更新信息参见:https://github.com/singleron-RD/AccuraCode







