分析软件
FocuSCOPE®单细胞血液肿瘤靶向基因突变检测数据分析
一、简介
新格元FocuSCOPE®系列产品创新性结合单细胞转录组技术与靶基因捕获技术,2021年相继推出FocuSCOPE®单细胞肺癌靶向基因突变检测试剂盒、FocuSCOPE®单细胞EB病毒靶向基因检测试剂盒。鉴于单细胞测序技术在血液病中的广泛应用,2022年5月推出FocuSCOPE®单细胞血液肿瘤靶向基因检测试剂盒,结合单细胞靶向技术鉴定血液肿瘤相关基因突变的细胞类型,研究不同驱动突变模式的细胞间基因表达差异。
FocuSCOPE®单细胞血液肿瘤靶向基因检测试剂盒,基于新格元的单细胞微流控系统分离细胞,独有的Barcode Beads设计的poly T探针用于捕获单个细胞的mRNA分子,靶向探针用于捕获血液肿瘤相关基因的热点区域。同时试剂盒内搭配反转录、cDNA富集、NGS建库的全流程试剂。可以快速、全面地完成单细胞转录组文库与靶基因富集文库构建。
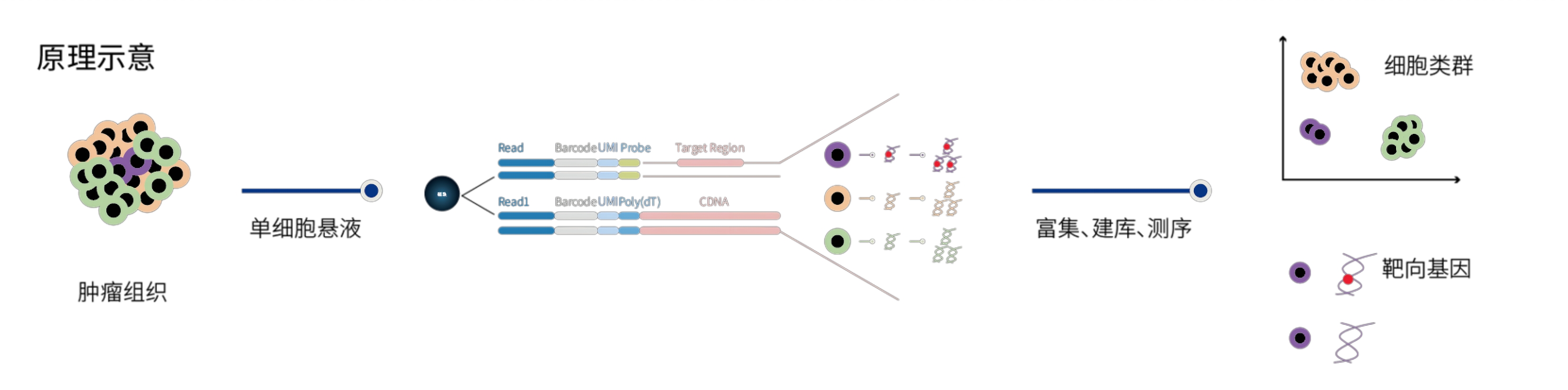
二、实验原理及流程
FocuSCOPE®单细胞血液肿瘤靶向基因突变检测试剂盒,可同时完成从组织样本运输、解离制备单细胞悬液、单细胞分离、mRNA以及肺癌靶基因的定向捕获和NGS测序文库构建整体流程。
三、产品优势
1. 突破位置限制
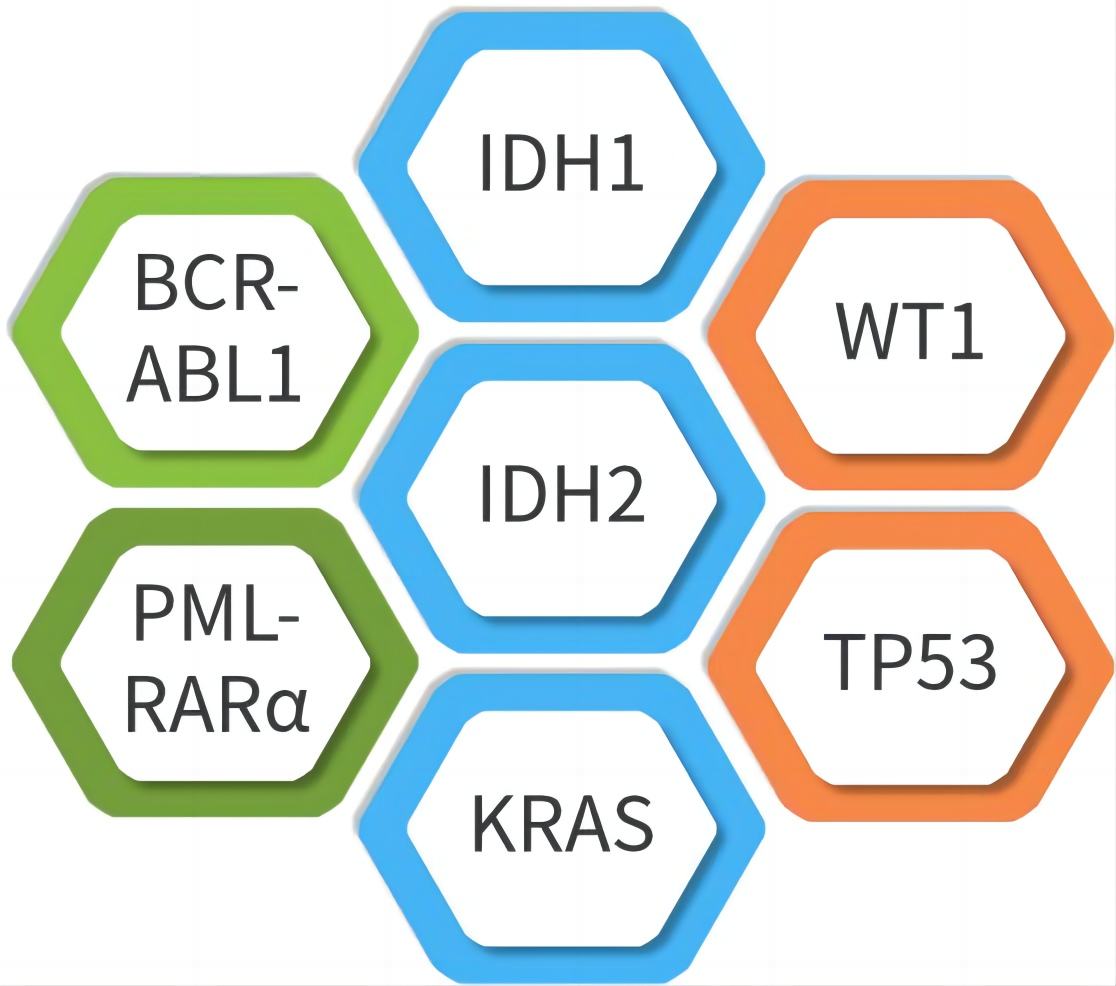
同时检出血液肿瘤靶药常见驱动基因突变和融合基因:
新型分子靶向药物的研发和应用已经广泛用于血液肿瘤治疗,如伊马替尼(Imatinib)可以显著改善慢性髓系白血病(CML)患者的生存期和预后,临床上可以根据患者是否存在BCR-ABL1融合基因来选择使用伊马替尼(Imatinib)治疗。
FocuSCOPE®单细胞血液肿瘤靶向基因突变检测试剂盒根据血液肿瘤靶药的5个常见驱动基因突变,以及2个融合基因进行靶向捕获和单细胞转录组同时检测。在单细胞层面检测药物敏感突变或耐药突变信息,为血液肿瘤微环境以及靶向基因突变提供新的解决方案。
2. 多技术结合
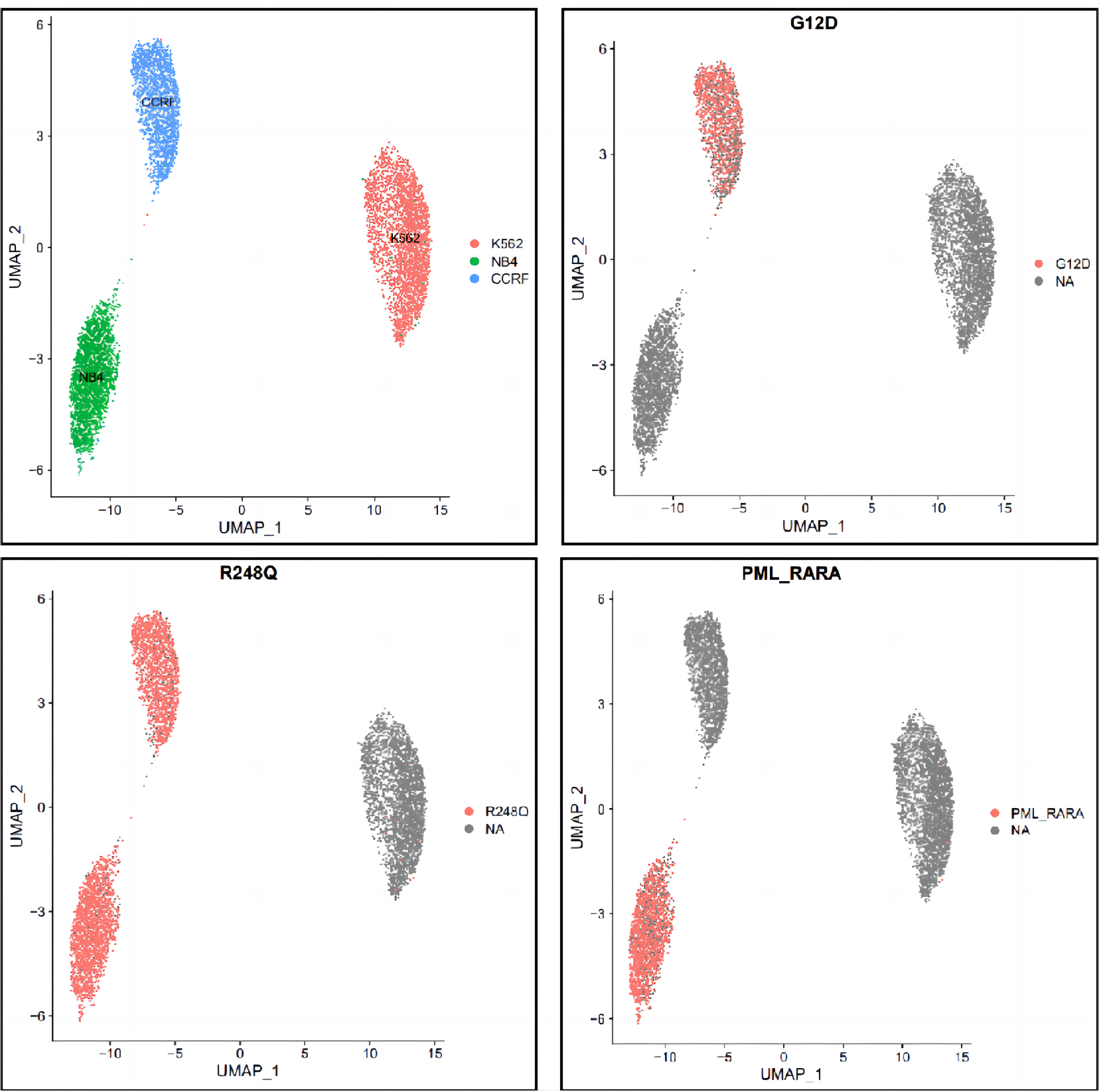
高通量单细胞转录组测序技术和基因突变靶向捕获技术:
使用NB4、CCRF、K562按照1:1:1投入构建混合细胞系,分别构建单细胞转录组文库和靶基因富集文库。从细胞分群图中可以看出3种类型细胞系可以清晰区分开,同时在CCRF细胞亚群中,可以检出血液肿瘤相关的SNV(KRAS:G12D;TP53:R175H、R248Q);在NB4细胞亚群中,血液肿瘤相关的SNV(KRAS:A18D;TP53:R248Q)和融合基因(PML-RARA)可以被很好检出。
3. 高灵敏捕获
全面检出较低细胞类型和基因突变信息:
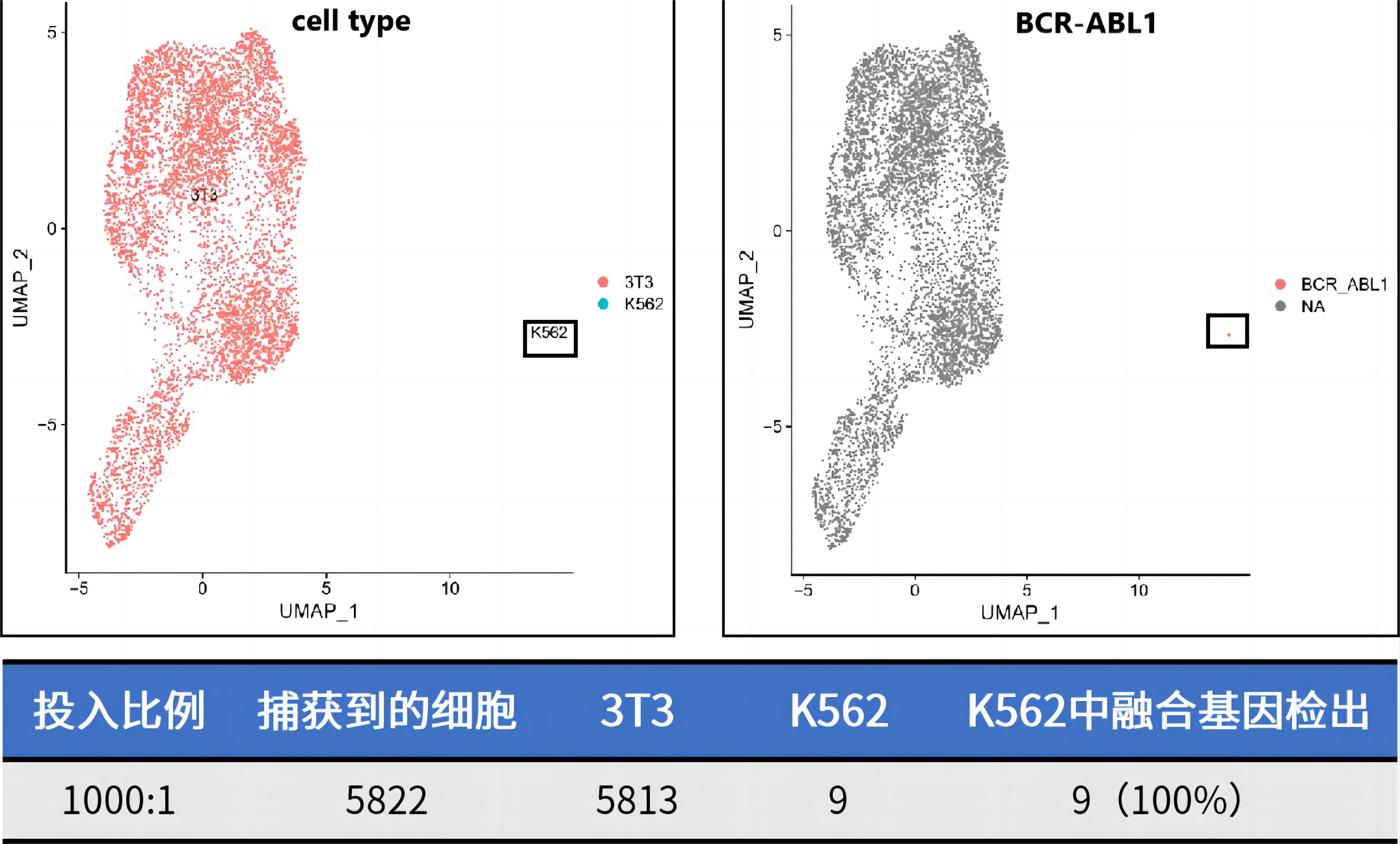
人细胞系K562含有血液肿瘤相关融合基因(BCR-ABL1),鼠细胞系3T3则不含,将3T3和K562细胞系按照1000:1比例混合,分别构建单细胞转录组文库和靶基因富集文库。从细胞分群图中可以看出2种类型细胞系可以清晰区分开,K562细胞系在投入和捕获细胞很少的情况下,也可以检出含有融合基因(BCR-ABL1)的细胞亚群。
四、应用场景
1. 血液肿瘤基因突变异质性研究
肿瘤高异质性与基因突变以及融合关系研究,探究基因突变对癌症相关基因表达的动态变化,为靶向治疗及其诊断提供了新的研究思路。
2. 血液疾病发生发展机制研究
血液肿瘤发生发展中的突变动态变化,比如白血病发生早期和晚期的差异基因突变,驱动突变在细胞层面的变化,引起细胞间互作模式改变。
3. 靶向药物筛选
加速对细胞突变分布和基因表达研究,为研究靶向药物作用机理、筛选新的靶向药提供单细胞数据,可以提高药物筛选的效率,促进新药的快速研制。
五、CeleScope fusion分析流程
FocuSCOPE生成的单细胞转录动态监测数据的基本流程。FocuSCOPE pipeline(单细胞FocuSCOPE®单细胞血液肿瘤靶向基因检测数据分析)包含8个主要指令,可以通过 celescope fusion {指令} --help 查看:
$ celescope fusion --help
usage: celescope fusion [-h] {mkref,sample,barcode,cutadapt,star_fusion,count_fusion,filter_fusion,analysis_fusion} ...
Single-cell fusion
positional arguments:
{mkref,sample,barcode,cutadapt,star_fusion,count_fusion,filter_fusion,analysis_fusion}
optional arguments:
-h, --help show this help message and exit
$ celescope -v
1.13.0下载测试数据与脚本
这些数据都是我们托管在GitHub上的测试数据,部分数据是人为生成的,仅供测试。
git clone https://github.com/singleron-RD/celescope_test_data.git
git clone https://github.com/singleron-RD/celescope_test_script.git
所有的软件DEMO测试数据我们已经在celescope rna的教程中下载过,这里可以看一下fusion数据的结构,我们看一下测试数据与脚本的结构:
# 测试数据
$ tree
.
├── fastqs
│ ├── fusion_1.fq.gz
│ └── fusion_2.fq.gz
└── match_dir
├── 05.count
│ └── fake_filtered_feature_bc_matrix
│ └── barcodes.tsv
└── 06.analysis
└── B1110-V7-2-R1V8-50_ZL_tsne_coord.tsv
# 测试脚本
$ tree
.
├── fusion_test.mapfile
└── run_shell.sh六、CeleScope fusion分析流程实操
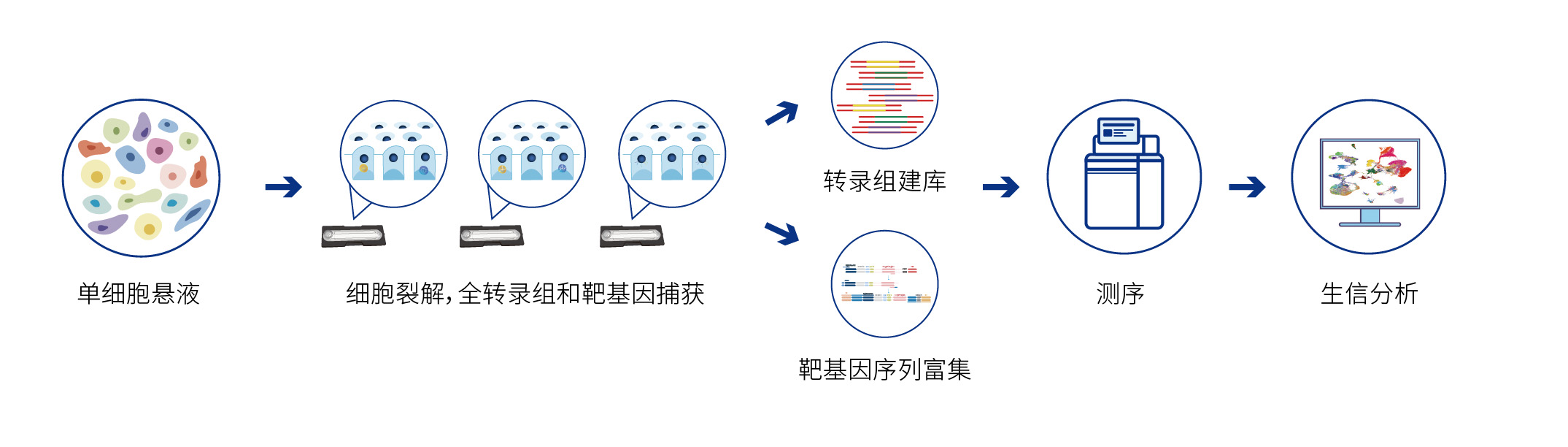
单细胞血液肿瘤靶向基因检测研究在实验过程中会构建一个单细胞转录组文库和单细胞血液肿瘤靶向基因序列富集。而单细胞血液肿瘤靶向基因检测可同时检测基因突变与融合基因,因此数据分析分为三个环节:(1)单细胞转录组分析;(2)单细胞血液肿瘤靶向基因突变检测分析;(3)单细胞血液肿瘤融合基因检测分析。
本篇文章内只介绍单细胞血液肿瘤融合基因检测分析流程,而GEXSCOPE®单细胞(核)转录组数据分析及FocuSCOPE®单细胞肺癌靶向基因突变数据分析的教程已在前期中介绍过。在分析之前我们先要激活我们CeleScope软件的运行环境,使用conda activate celescope命令进行激活。
1.用 mkref 指令创建一个参考基因组文件
-
mkref用于创建基因组参考目录 每个物种构建一次参考基因组即可,以后运行直接读取,不用多次构建。需要基因组序列文件与注释信息:fasta文件和gtf文件
第一步,下载血液肿瘤融合基因参考基因组序列文件首先我们下载我们在github上托管了测试数据,我们就可以获得血液肿瘤融合基因参考基因组序列文件。
mkdir test_dir
cd test_dir
git clone https://github.com/singleron-RD/CeleScope.git下载完成后,通过脚本命令cd celescope/data/fusion/blood_1/进入到blood_1文件目录中,查看目录里具体文件信息。
$ tree
.
├── BCR_ABL1_PML_RARA_all.fasta
├── BCR_ABL1_PML_RARA_all_pos.txt
└── mkref.sh
第二步, 使用mkref指令构建血液肿瘤融合基因参考基因组索引文件。
conda activate celescope
sh mkref.sh运行完成之后查看生成的索引文件目录。
$ tree
.
├── BCR_ABL1_PML_RARA_all.fasta
├── BCR_ABL1_PML_RARA_all_pos.txt
├── celescope_genome.config
├── chrLength.txt
├── chrNameLength.txt
├── chrName.txt
├── chrStart.txt
├── Genome
├── genomeParameters.txt
├── Log.out
├── mkref.sh
├── SA
└── SAindex2.用multi_fusion构建celescope fusion分析的shell脚本
一个是配置 mapfile文件--mapfile 是multi_fusion下的参数,需要提供一个制表符分隔 (tab-delimited) 的文本文件。mapfile的每一行代表双端(paired-end)fastq文件。
$ cat fusion_test.mapfile
fusion ../../celescope_test_data/fusion/fastqs test1 ../../celescope_test_data/fusion/match_dir第一列: fusion_fastq_ID:对应fusion_fastq文件的名称前缀 第二列: fusion_datapath:对应fusion_fastq文件的路径 第三列: fusion_name:对应质控报告的名称 第四列: 对应与其“配对的”单细胞转录组分析fake_match_dir路径
另一个是 shell 脚本文件:run_shell.sh
$ cat run_shell.sh
multi_fusion\
--mapfile ./fusion_test.mapfile\
--fusion_genomeDir /SGRNJ06/randd/public/genome/fusion/fusion/blood_1 \
--mod shell \
3.生成shell脚本
(1)运行刚编辑好的shell脚本run_shell.sh。
$sh run_shell.sh
(2)运行完成之后会生成一个名为shell的目录。
$ tree
.
├── fusion_test.mapfile
├── run_shell.sh
└── shellshell目录中会有一个以test1.sh命名的脚本,test1.sh脚本中的每行指令对应每一步分析(质控报告的每一部分数据)。
4.投递shell脚本
-
进入到shell目录后,就可以运行脚本test1.sh,然后在终端命令行中输入sh test1.sh。那么程序就会在当前的终端界面运行。但是,如果在当前的终端界面中进行运行,终端界面就不能关闭,也不能掉线,否则运行的程序就会中断。那么,为了避免这种情况发生,我们可以使用nohup将运行脚本投递后台运行,执行nohup sh test1.sh &,并生成一个nohup.out运行的日志文件。
$tree -L 1
.
├── nohup.out
├── test1
└── test1.sh-
如果对每一步做了什么感兴趣,可以单独运行查看,test1.sh里面是:
$ cat test1.sh
celescope fusion sample --outdir .//test1/00.sample --sample test1 --thread 4 --chemistry auto --fq1 /mnt/sdd/Singleron/bioinfo_test_data_and_script/celescope_test_data/fusion/fastqs/fusion_1.fq.gz
celescope fusion barcode --outdir .//test1/01.barcode --sample test1 --thread 4 --chemistry auto --lowNum 2 --fq1 /mnt/sdd/Singleron/bioinfo_test_data_and_script/celescope_test_data/fusion/fastqs/fusion_1.fq.gz --fq2 /mnt/sdd/Singleron/bioinfo_test_data_and_script/celescope_test_data/fusion/fastqs/fusion_2.fq.gz
celescope fusion cutadapt --outdir .//test1/02.cutadapt --sample test1 --thread 4 --minimum_length 20 --nextseq_trim 20 --overlap 10 --insert 150 --fq .//test1/01.barcode/test1_2.fq
celescope fusion star_fusion --outdir .//test1/03.star_fusion --sample test1 --thread 4 --outFilterMultimapNmax 1 --starMem 30 --fusion_genomeDir /mnt/sdd/Singleron/bioinfo_test_data_and_script/celescope_work_script/fusion/blood_1 --fq .//test1/02.cutadapt/test1_clean_2.fq
celescope fusion count_fusion --outdir .//test1/04.count_fusion --sample test1 --thread 4 --fusion_genomeDir /mnt/sdd/Singleron/bioinfo_test_data_and_script/celescope_work_script/fusion/blood_1 --flanking_base 5 --min_query_length 35 --capture_bam .//test1/03.star_fusion/test1_Aligned.sortedByCoord.out.bam --match_dir /mnt/sdd/Singleron/bioinfo_test_data_and_script/celescope_test_data/fusion/match_dir
celescope fusion filter_fusion --outdir .//test1/05.filter_fusion --sample test1 --thread 4 --read_threshold_method otsu --umi_threshold_method otsu --auto_coef 3 --otsu_log_base 10 --match_dir /mnt/sdd/Singleron/bioinfo_test_data_and_script/celescope_test_data/fusion/match_dir --raw_read_count_file .//test1/04.count_fusion/test1_raw_read_count.json
celescope fusion analysis_fusion --outdir .//test1/06.analysis_fusion --sample test1 --thread 4 --fusion_genomeDir /mnt/sdd/Singleron/bioinfo_test_data_and_script/celescope_work_script/fusion/blood_1 --match_dir /mnt/sdd/Singleron/bioinfo_test_data_and_script/celescope_test_data/fusion/match_dir --filter_umi_file .//test1/05.filter_fusion/test1_filtered_UMI.csv5.结果目录
$ tree
.
├── 00.sample
│ └── stat.txt
├── 01.barcode
│ ├── stat.txt
│ └── test1_2.fq
├── 02.cutadapt
│ ├── cutadapt.log
│ ├── stat.txt
│ └── test1_clean_2.fq
├── 03.star_fusion
│ ├── stat.txt
│ ├── test1_Aligned.out.bam
│ ├── test1_Aligned.sortedByCoord.out.bam
│ ├── test1_Aligned.sortedByCoord.out.bam.bai
│ ├── test1_Log.final.out
│ ├── test1_Log.out
│ ├── test1_Log.progress.out
│ └── test1_SJ.out.tab
├── 04.count_fusion
│ ├── stat.txt
│ ├── test1_raw_fusion.bam
│ └── test1_raw_read_count.json
├── 05.filter_fusion
│ ├── stat.txt
│ ├── test1_BCRe14_ABL1a2_read_otsu.png
│ ├── test1_corrected_read_count.json
│ ├── test1_filtered_read_count.json
│ └── test1_filtered_UMI.csv
├── 06.analysis_fusion
│ ├── stat.txt
│ ├── test1_BCRe14_ABL1a2_fusion.pdf
│ ├── test1_BCRe19_ABL1a2_fusion.pdf
│ ├── test1_BCRe1_ABL1a2_fusion.pdf
│ ├── test1_fusion_count.csv
│ ├── test1_PML6_RARA3_fusion.pdf
│ └── test1_UMI_tsne.csv
└── test1_report.html
当我们运行完以后,就可以得到一个单细胞血液肿瘤融合基因检测分析的网页报告。
-
质控报告的样本和软件的基本信息 -
数据质控信息 -
融合基因与基因组比对信息 -
细胞与基因定量情况 -
融合基因过滤后信息
以上就是完整的新格元单细胞血液肿瘤融合基因检测分析过程。
本文仅做软件安装测试使用,更多软件更新信息参见:https://github.com/singleron-RD/CeleScope







